REVIEW ARTICLE OPEN ACCESS
Autophagy-Driven Cancer Immunotherapy via Tumor Immune Ecosystem Remodeling
Yichen Liao1,2,3*, Xuxin Tan2*, Shuang Ren2, Chenyang Duan2#, Jun Hu3#
Received 2025 Sept 6
Accepted 2025 Nov 29
Epub ahead of print: December 2025
Published in issue 2026 Feb 15
Correspondence: Jun Hu - Email: hujun@tmmu.edu.cn
Chenyang Duan - Email: duanchenyang1991@cqmu.edu.cn
The author’s information is available at the end of the article.
© 2026 The Author(s). Published by GCINC Press. Open Access licensed under a Creative Commons Attribution 4.0 International License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. To view a copy: https://creativecommons.org/licenses/by/4.0/
Abstract
Autophagy is a conserved catabolic pathway essential for maintaining cellular integrity, recycling damaged organelles, and supporting metabolic adaptation during stress. Beyond its homeostatic functions, aberrant autophagy plays a critical role in cancer initiation and progression. Once viewed primarily as a tumor-suppressive mechanism linked to programmed cell death, autophagy is now recognized as a highly context-dependent process that can either inhibit or facilitate tumor development. Growing evidence demonstrates that autophagy regulates multiple cancer hallmarks, including metastasis, sustained proliferation, therapeutic resistance, and immune regulation. In this review, we explore how autophagy intersects with the immune system to remodel the tumor microenvironment (TME), highlighting its dual and often paradoxical roles. Autophagy shapes the activation, differentiation, and effector functions of both innate and adaptive immune cells, enhancing antitumor immunity while also promoting immune evasion. Major TME constituents, such as tumor-associated macrophages, cancer-associated fibroblasts, dendritic cells, natural killer cells, and cytotoxic T lymphocytes, undergo autophagy-dependent reprogramming, particularly in response to hypoxia, nutrient stress, and inflammatory cues. Notably, autophagy-driven immunogenic cell death has emerged as a promising avenue to augment cancer immunotherapies, including immune checkpoint inhibitors and adoptive cell therapies. Recent preclinical and clinical advances targeting autophagy pathways underscore new therapeutic opportunities and position autophagy modulators as emerging immunopharmacological agents. Elucidating how autophagy-mediated immune remodeling shapes the TME may enable the development of next-generation precision cancer therapies.
Keywords: Autophagy, Cancer Immunotherapy, Tumor Microenvironment, Immunogenic Cell Death (ICD), Immune Reprogramming, Tumor Immune Evasion.
1. Introduction
Cancer is a leading cause of death. Despite advances in therapy, it remains incurable. The tumor microenvironment (TME) is a dynamic ecosystem containing cancerous and non-cancerous cells and is increasingly recognized as a major regulator of tumorigenesis (1-3). The TME influences proliferation, metastasis, and drug resistance (4-6). Stromal cells, such as fibroblasts and mesenchymal cells, constitute this environment. Cancer-associated fibroblasts (CAFs) in the TME enhance carcinogenesis and angiogenesis by secreting growth factors and cytokines and by regulating extracellular matrix (ECM) components (7-10). The TME also includes immune cells, such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs), each of which influences tumorigenesis. M2-type macrophages create an immunosuppressive environment (11-13). MDSCs and Tregs suppress T cells and natural killer (NK) cells, enabling immune evasion. Hypoxic conditions in the TME trigger angiogenesis and VEGF upregulation, increasing tumor aggressiveness (14-16). Matrix metalloproteinases (MMPs) promote ECM degradation, facilitating metastasis and invasion (17-19). Signaling molecules, including cytokines, chemokines, and growth factors, drive tumorigenesis, angiogenesis, and epithelial-mesenchymal transition (EMT). Understanding mechanisms of TME remodeling, such as autophagy, is critical (Figure 1).
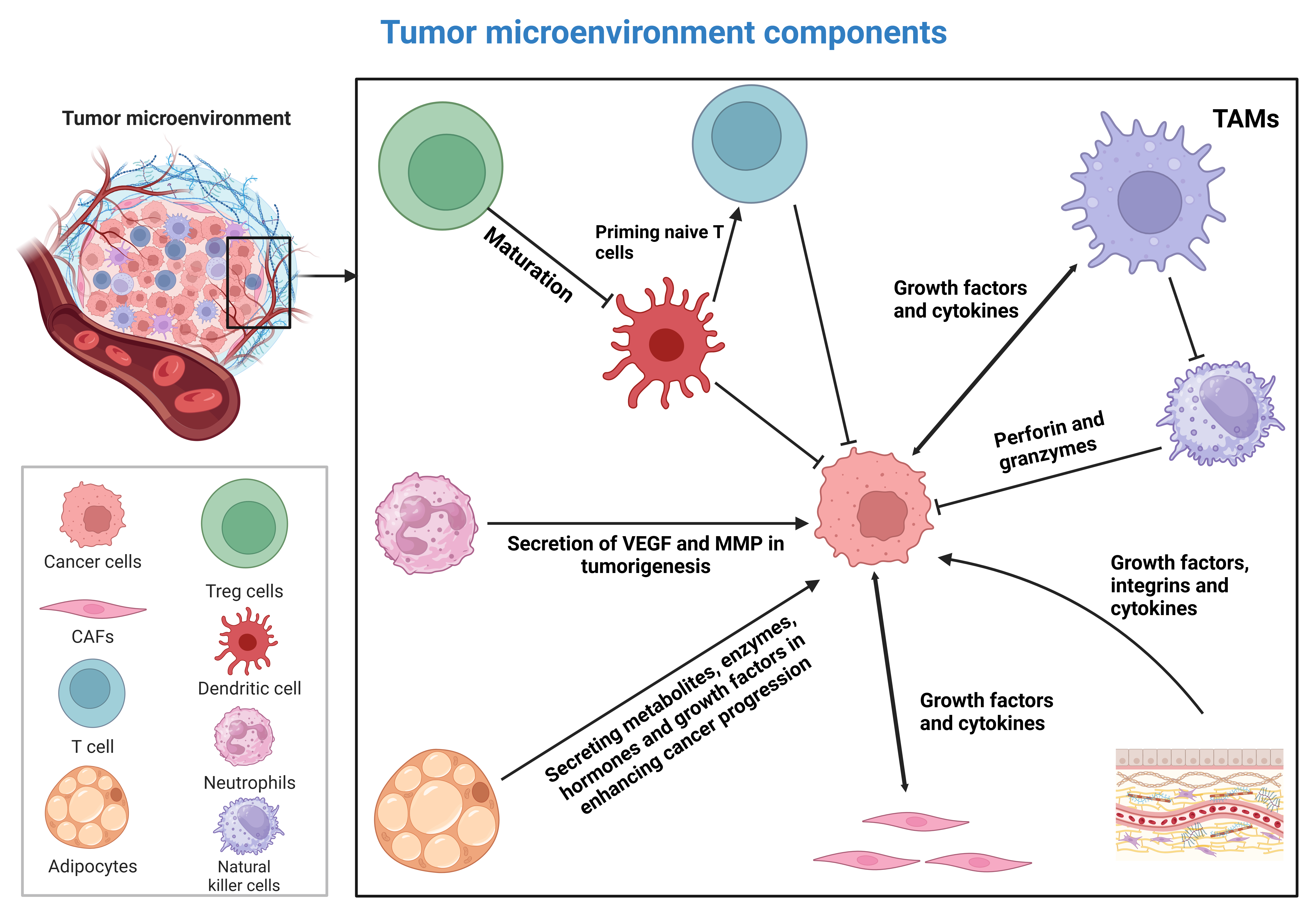
Figure 1: A schematic representation of the tumor microenvironment (20). The maturation of dendritic cells is required for priming naïve T cells in cancer immunotherapy. However, this process is suppressed by regulatory T cells. Tumor-associated macrophages can suppress the function of natural killer cells. When polarized into the M2 phenotype, they create an immunosuppressive microenvironment. The extracellular matrix secretes growth factors, integrins, and cytokines that enhance tumorigenesis. Tumor cells induce cancer-associated fibroblasts. These fibroblasts then secrete growth factors and cytokines to enhance cancer progression. Adipocytes and neutrophils are additional components of the tumor microenvironment. They enhance tumorigenesis by secreting VEGF, MMPs, enzymes, and metabolites. This figure was created using BioRender.
Conventional therapeutics have significant challenges in treating cancer. As a result, combination therapies and newer approaches, such as immunotherapy, are being pursued to eliminate cancer. However, tumor microenvironment (TME) remodeling and other factors promote immune evasion (21). The TME supports immune evasion through a dense extracellular matrix (ECM) and abnormal vasculature. Thus, regulating the TME is essential for reversing immune evasion (22, 23). Tumor cells can downregulate MHC class I, reducing antigen presentation, impairing T cell function, and promoting immune evasion (24-26). The immunosuppressive environment is further mediated by M2-polarized macrophages (27), Tregs (28), and MDSCs (29). Cytokines such as IL-10 and TGF-β impair immune cell function and promote immune evasion (30, 31). Metabolites such as adenosine (32) and indoleamine 2,3-dioxygenase (IDO) (33) also contribute to the immunosuppressive TME. Thus, recognizing and targeting these factors is crucial in cancer immunotherapy.
This review will focus on autophagy’s regulation of TME components and its effects on immune responses. First, the mechanisms of autophagy and their regulation are highlighted. Then, how autophagy regulates the innate and adaptive immune systems is described. The potential of autophagy to modulate immune evasion is also discussed. The role of autophagy in regulating immunogenic cell death and its changes under hypoxic conditions are covered. Finally, the specific role of autophagy in regulating TME components is examined to highlight its potential for TME remodeling and modulation of immune evasion.
2. Autophagy machinery: Basics and principles
In recent years, the importance of cell death mechanisms has been highlighted. They are essential for developmental processes, tissue homeostasis, and the elimination of damaged cells. These mechanisms have drawn interest in human cancers (34-36). Among them, the role of autophagy in tumorigenesis has been extensively studied. Autophagy is a programmed cell death pathway comprising macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy (hereafter called autophagy) is the primary type. Although it is known as a cell death pathway, autophagy plays a dual role in cancer, acting either tumor-promoting or tumor-suppressing. It can regulate several cancer hallmarks (37-41). Basic levels of autophagy are maintained in cells to prevent the accumulation of damaged organelles, toxic macromolecules, and unfolded proteins. However, certain conditions, such as starvation, can stimulate autophagy. Autophagy has four steps: initiation, nucleation, elongation, and maturation. The mechanistic target of rapamycin complex 1 (mTORC1) strictly regulates the initiation of autophagy. During nutrient-rich conditions, mTORC1 suppresses autophagy by downregulating the unc-51-like autophagy activating kinase 1 (ULK1) complex. When nutrients are deficient, mTORC1 becomes inactive. This allows ULK1 to induce autophagy. Upon induction, ULK1 participates in the phosphorylation of the class III PI3K complex. This complex, comprised of Beclin-1, VPS34, and ATG14, produces PI3P on the phagophore membrane and recruits other ATG proteins. Two ubiquitin-like conjugation systems contribute to the elongation of the phagophore membrane. The ATG12-ATG5-ATG16L1 complex acts as an E3 ligase, accelerating the conjugation of LC3 to PE to form LC3-II. LC3-II then associates with the autophagosome membrane, promoting its expansion and cargo selection. Maturation involves the fusion of autophagosomes and lysosomes to generate autolysosomes. Proteins like SNAREs and LAMP1/2 contribute to this step. Finally, the cargo in autophagosomes is degraded by lysosomal enzymes. More detailed information on autophagy is available in the reviews (38, 40, 42-45) and in Figure 2.
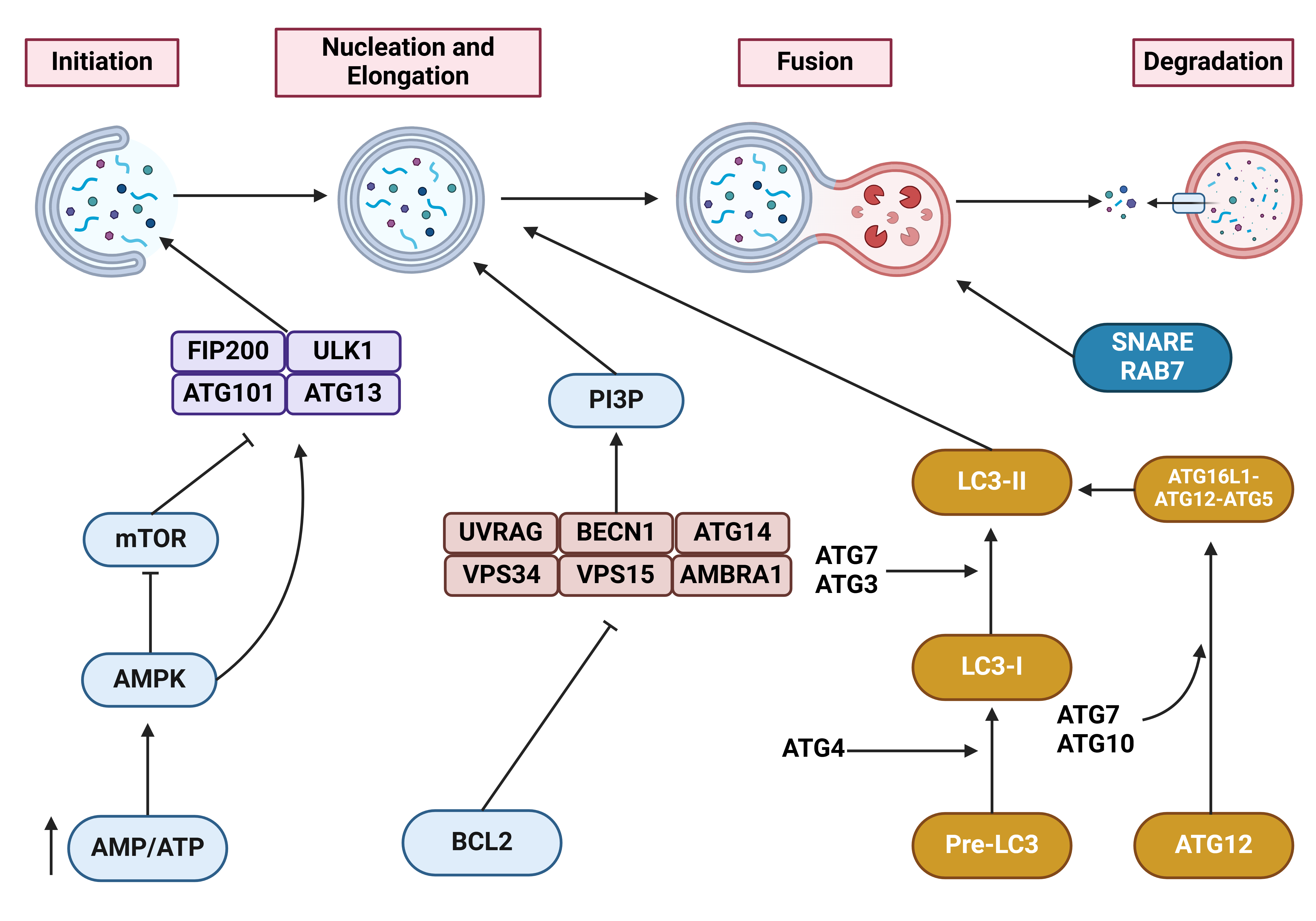
Figure 2: A detailed representation of the autophagy mechanism (46). When AMP levels increase and surpass ATP levels, AMPK is upregulated to suppress mTOR. Then, the induction of FIP200-ULK1-ATG101-ATG13 complex initiates autophagy. In another step, BCL2 suppresses UVRAG, BECN1, ATG14, VPS34, VPS15 and AMBRA1. This complex can induce PI3P-mediated autophagy nucleation. Moreover, ATG4 mediates the conversion of pre-LC3 to LC3-I. ATG7 and ATG3 mediate the conversion of LC3-I to LC3-II, thereby promoting autophagosome maturation. SNARE and Rab7 are among the proteins vital to autophagosome-lysosome fusion. This figure was created using BioRender.
The multifaceted role of autophagy in cancer has been challenging, as this mechanism can exert both pro- and antitumor effects, making it difficult to develop autophagy inhibitors or inducers. Furthermore, autophagy is a potent regulator of drug resistance and affects the growth and invasion of cancer cells (47-49). In addition to drug resistance, autophagy can control radioresistance. Notably, Hexokinase-2 has been shown to promote AIMP2 degradation via autophagy, thereby increasing radioresistance in hepatocellular carcinoma (50). The studies have focused on the regulatory mechanisms of autophagy in human cancers. The upregulation of HSP70 is found in pancreatic cancer, and it increases cancer metastasis. Downregulation of HSP70 can impair mitochondrial dynamics and promote apoptosis.
HSP70 downregulation increases AMPK levels, thereby promoting Beclin-1 phosphorylation and autophagy induction (51). Moreover, epigenetic regulation of autophagy can occur in cancers. LncRNA PRBC accelerates breast cancer progression by interacting with PABPC1, promoting its nuclear translocation and inducing autophagy (52). CSNK2 can suppress autophagy by inducing FLN-NHL-containing TRIM proteins (53), and this strategy can be used to exploit autophagy's protective function to impair carcinogenesis. The proper function of some organelles in cells, such as mitochondria, also depends on autophagy. The suppression of autophagy can trigger mitochondrial dysfunction in pancreatic cancer by reducing SDHB levels (54). Moreover, autophagy can regulate other cell death mechanisms in cancer, such as ferroptosis (55). Given the critical role of autophagy in human cancers, various autophagy modulators, including natural products, small molecules, and nanostructures, have been developed to control autophagy (56-62). One of the most recent advances is the recognition of the role of autophagy in cancer immunotherapy. For example, the suppression of autophagy in CAFs has been shown to impair adaptive immune resistance in pancreatic cancer and enhance the response to immunotherapy (63). Hence, the following sections will focus on the role of autophagy in regulating immune responses, immune evasion, and TME remodeling.
3. Autophagy in the innate immune system
Adaptive and innate immune responses are responsible for cancer surveillance (64). Autophagy can activate innate immune receptors, such as TLRs and NLRs, thereby accelerating effector responses. The induction of innate immune receptors, including TLRs and NLRs, can be regulated by autophagy (65). TLR2-induced autophagy can upregulate JNK and ERK, thereby enhancing innate immune responses (66, 67). TLR7 can contribute to the elimination of intracellular residues through enhancing ATG5 and Beclin-1 levels in a MyD88-related manner (68). Another approach is the TLR4 signaling pathway, which induces the RIP1/MAPK axis to mediate autophagy (69). However, depending on the condition, the factors responsible for induction differ. TICAM1/TRIG is essential for the function of TLR4 and TLR3 in autophagy activation by lipopolysaccharides (LPS) and polyinosinic-polycytidylic acid (poly (I: C)), respectively, which facilitates TRAF6 ubiquitination and subsequent induction of MAPK and NF-KB. This elevates cytokine production, thereby increasing tumor cell migration (70). Moreover, TLRs are not the only factors mediating autophagy. The DNA damage-regulated autophagy modulator 1 (DRAM1) enhances pathogen identification via the TLR-MYD88-NF-κB innate immune signaling pathway, triggering selective autophagy (71). Although TLRs detect microbes on the cell surface, NOD1 and NOD2 can also detect cytosolic pathogens via iE-DAP and MDP, respectively. These factors can enhance the production of pro-inflammatory factors and mediate the effects of immunosuppressive cytokines by upregulating NF-κB and MAPK signaling. NOD1 and NOD2 can increase ATG16L1 levels, thereby enhancing autophagosome formation (72).
4. Autophagy in the adaptive immune system
In the adaptive immune system, autophagy regulates antigen presentation, MHC molecules, and T and B cells (65). The process of taking up and processing exogenous antigens and presenting them to antigen-specific T cells is known as cross-presentation, which is mediated by antigen-presenting cells (APCs) and is an essential step for the adaptive immune system (73). Autophagy is critical for antigen presentation and can deliver antigens to MHC class I molecules, thereby facilitating cross-presentation (74). Suppression of autophagy increases the number of intracellular bacteria (75). TNF-α-induced autophagy is vital for the processing and presentation of mitochondrial antigens by MHC-I molecules (76). Autophagy is essential for MHC-I antigen expression, but its role in regulating tumor immunity differs. At the early stage of melanoma, autophagy can promote CTL-mediated cytolysis of melanoma cells, whereas it disrupts cytolysis in advanced stages (77).
Furthermore, the autophagy mechanism is vital for maintaining T cell homeostasis and for inducing T cells. Autophagy can be stimulated by TCR activation and can preserve T cell organelle homeostasis (78, 79). Autophagy-related factors are responsible for inducing T cells. Notably, knockdown of ATG3, ATG5, ATG7, and Beclin-1, as well as VPS34, can suppress autophagy and impair T cell homeostasis by regulating mitochondrial quality control (78, 80, 81). Autophagy has also been shown to regulate helper T lymphocyte (HTL) function and to mediate anti-cancer immune responses against tumors expressing c-Met (82). In addition, B-cell development and survival can be regulated by autophagy. The survival of autoreactive B cells can be increased by autophagy (83). The B cell development, B cell maintenance, and plasma cell development depend on ATG5 expression (84, 85). DRibbles, as tumor-derived autophagosomes, have been shown to stimulate B cells. Macrophages can mediate TLR4 and MyD88 signaling to induce DRibbles-mediated B cell function (86).
5. Autophagy and immune evasion in human cancers
In recent years, a special focus has been on the mechanisms underlying immune evasion in human cancers. The importance of immunotherapy in cancer has been underscored, and the molecular pathways that regulate immune evasion have been highlighted. In the case of KEAP1 mutation, Nrf2 upregulation mediates immune evasion in lung cancer (87). For immunotherapy, it is necessary to mediate an “eat-me” signal, and in this context, A20 increases STC1 expression to promote immune evasion in colorectal tumors (88). Another factor contributing to immune evasion by cancer cells is the overexpression of PD-L1. Notably, ANXA1 overexpression can bind PARP1 and enhance STAT3 levels, thereby promoting PD-L1 expression and immune evasion (89). The increase in PD-L1 expression can also be mediated by c-Myc, which induces immune evasion (90). Immune evasion has been reported in various human cancers, including bladder cancer (91), colon cancer (92), and breast cancer (93), among others. In this section, the role of autophagy in regulating immune evasion is described.
Autophagy helps pancreatic cancer evade the immune system by breaking down MHC-I (94). MHC-I molecules in pancreatic cancer are degraded via autophagy, with NBR1 involvement, leading to decreased cell-surface expression. Blocking autophagy increases MHC-I levels, thereby enhancing antigen presentation and T-cell responses, leading to reduced tumor growth in mice. Depleting CD8+ T cells or lowering MHC-I expression reverses the effects of autophagy inhibition. Enhancing anti-tumor immune response by combining autophagy inhibition with dual ICB therapy. These findings indicate that enhanced autophagy or lysosomal function contributes to immune evasion in pancreatic cancer, supporting the use of autophagy suppression and combination ICB therapy for disease treatment (94). In addition, immune evasion in pancreatic cancer is enhanced by the selective autophagy of MHC-I (95). This study discovered a new mechanism by which NBR1 facilitates immune evasion by pancreatic cancer cells through selective macroautophagy of MHC-I. Crucially, blocking autophagy or lysosomal function increases MHC-I expression, strengthening T cell-mediated tumor immunity and boosting the effectiveness of immune checkpoint blockade in transplanted tumor models in mice with similar genetic backgrounds (95).
Moreover, NLRC5-mediated selective autophagy promotes immune evasion in endometrial cancer. In vitro, autophagy suppresses NLRC5 and NLRC5-induced MHC-I gene expression. It is worth mentioning that the autophagy protein MAP1LC3/LC3 interacts with NLRC5 to block the NLRC5-mediated MHC-I antigen presentation pathway in endometrial cancer. The findings reveal a new role for the autophagy protein LC3 in controlling NLRC5-dependent MHC-I antigen presentation in endometrial cancer, suggesting a possible immunotherapy strategy for EC patients by suppressing LC3 and activating NLRC5 (96).
The activation of 5-hydroxytryptamine1A (5-HT) receptors on cancer cells promotes immune evasion in patients with lung adenocarcinoma and depression via the autophagy/pSTAT3 pathway (97). Patients with depression who have lung adenocarcinomas show higher levels of 5-HT, increased 5-HT1R expression, and decreased survival compared to patients without depression. Additionally, 5-HT1aR plays an essential role in increasing the number of CD4+CD25+Foxp3+ Treg cells and reducing the Th1/Th2 ratio, suggesting possible immune dysfunction. Furthermore, the presence of 5-HT1aR on cancer cells was inversely linked to CTL function in both peripheral blood and tumor-infiltrating lymphocytes. When someone is feeling down, 5-HT1aR activates p-STAT3 and autophagy, leading to the activation of programmed death ligand-1, which creates a suppressive immune environment.
Additionally, reduced overall survival was associated with increased activation of 5HT1aR and a heightened tumor autophagy/p-STAT3 axis in both the mouse model and lung adenocarcinoma patients (97). C-MYC suppressed ferroptosis and enhanced immune escape in ovarian cancer cells by regulating ferritin autophagy through NCOA4 (98). The upregulation of C-MYC and downregulation of NCOA4 were linked to the severity of the cancer. C-MYC was found to bind NCOA4 mRNA, decreasing NCOA4 levels and blocking ferroptosis, thereby lowering ROS levels. This led to increased growth and spread of cancerous cells. Furthermore, C-MYC blocked HMGB1 release via the NCOA4 axis, thereby promoting cancer development and evading immune response. In general, C-MYC helps regulate pathways that promote the progression and spread of ovarian cancer (98).
6. Hypoxia-mediated autophagy regulation: Perspectives on cancer immunotherapy
In the TME, specific conditions can alter the tumor cell progression pathway. Due to competition for nutrients and oxygen and the production of waste products, hypoxia is among the most common conditions in the TME. The presence of hypoxia can affect the underlying molecular pathways in regulating the progression of human cancers. Notably, hypoxia induces angiogenesis and immunosuppression in cancer (99). Moreover, hypoxia induces tumor cells to secrete exosomes, thereby affecting tumor growth and invasion (100). The upregulation of SLC2A12 during hypoxia can impair ferroptosis, enhancing the proliferation and invasion of ovarian tumors (101). Given these facts, this section focuses on the role of hypoxia in regulating autophagy, offering further insights into cancer immunotherapy.
FUS-circTBC1D14 stress granules induced by hypoxia stimulate autophagy in breast tumor (102). Hypoxia can trigger the formation of stress granules in the cytoplasm via FUS-circTBC1D14, with PRMT1 involvement. Subsequently, circTBC1D14 enhances PRMT1 stability by preventing K48-regulated polyubiquitination, thereby increasing PRMT1 levels. Furthermore, FUS-circTBC1D14 SGs can start a series of SG-bound proteins that regulate the removal of SGs by bringing in LAMP1 and boosting autophagy flux associated with lysosomes. This helps maintain cellular balance and supports tumor progression (102). Moreover, PAK1 acetylation induced by hypoxia extends autophagy and facilitates brain tumor growth by phosphorylating ATG5 (103). PAK1 is overexpressed and promotes glioblastoma growth by enhancing autophagy. Hypoxia induces ELP3-mediated acetylation of PAK1 at K420, thereby enhancing its function and promoting phosphorylation of ATG5 at T101. This hinders ATG5 degradation and enhances autophagosome formation, both of which are essential for glioblastoma growth. Using shRNA or FRAX597 to block PAK1 suppresses autophagy and tumor growth, whereas SIRT1-mediated PAK1-deacetylation impedes these activities. In glioblastoma patients, there is a clinical correlation between PAK1 acetylation and ATG5 phosphorylation. The research shows that PAK1 is involved in autophagy induced by low oxygen levels and in glioblastoma growth, suggesting it could be a promising target for glioblastoma treatment. Acetylation alterations and PAK1 kinase activity are essential for initiating autophagy and maintaining glioblastoma growth (103).
Hypoxia-activated PVT1 enhances lung cancer resistance to cisplatin through autophagy by regulating the PVT1/miR-140-3p/ATG5 pathway (104). PVT1 is abundantly expressed in lung cancer and in cell lines, and sHIF-1α regulates its expression. HIF-1α can bind to the PVT1 promoter and regulate its expression. PVT1 contributed to hypoxia-induced chemoresistance through the autophagy signaling pathway via the PVT1/miR-140-3p/ATG5 axis, leading to increased viability and reduced apoptosis (104). Furthermore, BNIP3 enhances pancreatic cancer cell migration and growth by regulating autophagy under hypoxic conditions (105). Under hypoxic conditions, BNIP3 expression increased, promoting the migration and growth of CFPAC-1 and Panc1 cells. Increasing BNIP3 levels was crucial for triggering autophagy, whereas silencing Atg5 and Atg7 with siRNA inhibited autophagy and hypoxia-induced cell migration and proliferation. Moreover, inhibition of ERK1/2 MAPK signaling by PD98058 led to a notable decrease in BNIP3 expression, autophagy induction, and the movement and growth of CFPAC-1 and Panc1 cells under low-oxygen conditions (105). Notably, natural compounds can regulate autophagy under hypoxic conditions. Apigenin (APG) triggers autophagy and cell death in gastric cancer cells under low-oxygen conditions by targeting EZH2 (106). APG enhanced autophagic cell death in gastric cancer by upregulating ATG5, LC3-II, and phosphorylation of AMPK and ULK1 while decreasing p-mTOR and p62 levels. APG triggers autophagic cell death by activating the PERK pathway, suggesting a response to endoplasmic reticulum (ER) stress. Blocking ER stress reduced APG-induced autophagy and promoted prolonged cell viability, suggesting that autophagic cell death was prevented. APG causes cell death related to ER stress and autophagy by inhibiting HIF-1α and Ezh2 in both normoxic and hypoxic conditions. Hence, APG induces autophagic cell death in GC cells by inhibiting HIF-1α and Ezh2 under hypoxic conditions (106). Moreover, hypoxia induces autophagy by increasing lysosomal protein translation in human colon cancer cells (107). Pathway enrichment analysis indicated that several up-regulated genes are involved in lysosomal, glycan, and lipid metabolism; antigen presentation; cell adhesion; and modification of the extracellular matrix and cytoskeleton. Most of the down-regulated genes play roles in apoptosis, ubiquitin-mediated proteolysis, and oxidative phosphorylation. In HCT116 cells, hypoxia triggers lysosomal autophagy and mitochondrial dysfunction via translational regulation. HCT116 cells experience hypoxia-induced mitochondrial autophagy through the involvement of numerous translation factors and mTOR kinase activity (107). Given the roles of autophagy in cancer immunotherapy (108, 109) and of hypoxia in immunosuppression (110, 111), the interaction between hypoxia and autophagy warrants further analysis in immune regulation.
7. Autophagy-accelerated immunogenic cell death: Perspectives on cancer immunotherapy
A new form of regulated cell death, immunogenic cell death (ICD), can mediate the release of damage-associated molecular patterns (DAMPs) and tumor-associated antigens (112). This enhances dendritic cell (DC) maturation and promotes the infiltration of cytotoxic T lymphocytes (CTLs). This is vital for alleviating immunosuppressive TME and promoting sensitivity to immunotherapy. DAMPs have been identified as calreticulin (CRT) on the tumor surface, high-mobility group box 1 (HMGB1), secreted ATP, and type I interferon (IFN) (113). Notably, the presence of CRT on the surface of cancer cells can bind to CD91 receptors on antigen-presenting cells (APCs), thereby providing an “eat-me” signal. This enhances APCs' ability to identify tumor antigens, thereby inducing the innate and adaptive immune systems (114). In recent years, ICD induction has emerged as a promising approach to cancer treatment (115-120). This section focuses on the interaction between autophagy and ICD in cancer therapy.
Brucine-induced lysosomal dysfunction impairs autophagy, leading to ICD (121). Brucine, a small molecule commonly used in traditional Chinese medicine, suppressed autophagy. Brucine also caused cell stress and led to characteristics of ICD, such as CRT exposure and release of HMGB1. Brucine inhibited the self-degradation process and modulated the ERK1/2-mTOR-p70S6K signaling cascade via feedback regulation. Reduced autophagy contributed to the development of Brucine-induced ICD, with blocking Atg5 leading to a significant decrease in Brucine-induced CRT presentation and HMGB1 release (121). Norcantharidin (NCTD) promotes autophagy in acidic conditions to induce ICD of bladder cancer (122). NCTD successfully eradicates bladder cancer in acidic environments and induces ICD, bolstering anti-tumor immune response. Treatment with NCTD increased surface CRT on cancer cells and facilitated DC maturation. Autophagy was discovered to be involved in NCTD-induced ICD. DC maturation was different between acidic and physiological pH conditions. NCTD therapy increased tumor-infiltrating T lymphocytes and enhanced survival without tumors after mice were vaccinated with NCTD-treated bladder cancer cells (122).
Furthermore, nanoparticles that enhance autophagy trigger ICD when combined with anti-PD-1/PD-L1 for the immunotherapy of residual tumors following radiofrequency ablation (123). SBZP nanoparticles (comprised of ZIF-8 structures) restricted the growth of cancer cells in the presence of heat stress by triggering apoptosis through a mechanism that depends on autophagy. The SZP nanoparticles also boosted autophagy in cancer cells and stimulated ICD, thereby enhancing DC maturation. Vaccinating with heat + SZP treatment successfully halted tumor growth and created lasting immunity. SBZP nanoparticles enhanced immune cell death, triggered immune responses against tumors, and inhibited tumor growth, indicating a promising strategy for tackling remaining tumors after radiofrequency ablation (123). Notably, the drugs can also be used for the induction of ICD. Thioridazine (THD) has been shown to trigger ICD in colorectal cancer by activating the eIF2α/ATF4/CHOP and secretory autophagy pathways (124). THD triggers ER stress by activating the eIF2α/ATF4/CHOP pathway and promoting the accumulation of secretory autophagosomes, leading to ICD. Moreover, when combined with oxaliplatin (OXA), THD significantly activated ICD, halting tumor progression in a mouse model (124).
Furthermore, ROS-induced autophagy in cancer cells helps these cells evade factors that trigger ICD (125). Inhibiting autophagy in cancer cells through ATG5 knockdown did not affect ATP secretion following Hyp-PDT. Cancer cells with reduced autophagy showed increased ecto-CALR induction after Hyp-PDT, which was closely linked to their inability to remove oxidatively damaged proteins. Additionally, the suppression of autophagy in Hyp-PDT-treated cancer cells enhanced their capacity to promote DC maturation, IL-6 production, and the proliferation of CD4+ or CD8+ T cells, as well as increased IFNG production (125). Autophagy-dependent ICD is triggered in lung cancer cells by the oncolytic Newcastle disease virus (126). Examination of lung cancer cells infected with NDV revealed an increase in live cells upon exposure to CRT. There was an increase in CRT localization on cell membranes after infection. HMGB1 and HSP70/90 were secreted during NDV infection. Pan-caspase inhibitors and necrosis inhibitors did not affect the release of ICD determinants. Suppression of autophagy-related genes prevented activation of ICD determinants. In a model of lung cancer using xenografts, tumor growth was suppressed by treatment with NDV-infected cell supernatants. In general, oncolytic NDV triggers strong ICD in lung cancer cells, with autophagy contributing to this mechanism (126). Therefore, ICD is beneficial in cancer therapy by maturing DCs to induce T-cell responses. Hence, induction of pro-death autophagy can accelerate ICD in cancer immunotherapy (Figure 3).
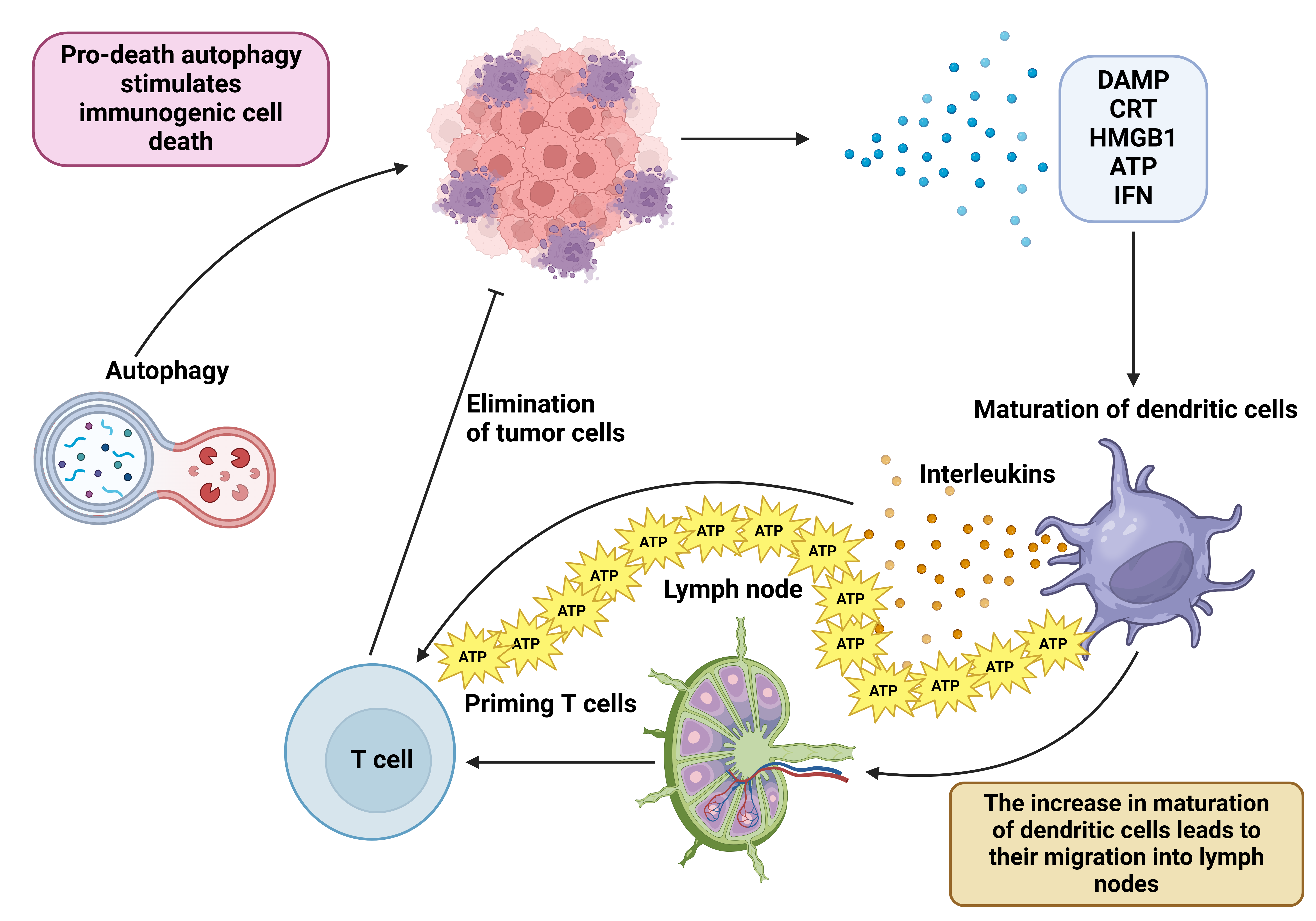
Figure 3: Autophagy and immunogenic cell death interaction in cancer immunotherapy. Autophagy with cytotoxic activity can mediate immunogenic cell death. Subsequently, the release of DAMP, CRT, HMGB1, ATP, and IFN promotes dendritic cell maturation and mediates their migration into lymph nodes, thereby priming naïve T cells in cancer immunotherapy. This figure was created using BioRender.
7.1 Autophagy, cytokines, interferons, and inflammatory factors
Increasing evidence indicates that autophagy interacts with cytokines and other factors in human cancers (127). IL-1 is a pro-inflammatory factor that can reduce COX-1, IκB, and JNK, thereby promoting tumorigenesis. IL-1 suppression has been shown to reduce cancer progression (128). In most cases, autophagy reduces IL-1β levels (129). Notably, IL-1α and IL-1β can induce autophagy, highlighting a negative feedback loop (130). IL-1β secretion can be diminished by stimulating autophagy, thereby reducing T cell function and cytokine production (131). The absence of autophagy in macrophages can increase IL-1 levels, thereby promoting tumorigenesis via the ROS/NF-κB axis (132). IL-2 has been shown to enhance levels of ATG5, HMGB1, and Beclin-1, thereby inducing autophagy (133). Moreover, IL-6 increases STAT3 levels and downregulates LC3-II and Beclin-1, thereby suppressing autophagy (134). A similar pathway is followed by IL-10, which stimulates the STAT3 and PI3K/Akt/mTORC1 axes to impair autophagy (135). IL-4, IL-13, and IL-10 are Th2 cytokines that can impair autophagy (136). Similar to cytokines, interferons (IFN) can mediate autophagy through increasing JAK/STAT levels (137). IFN-γ has been shown to stimulate autophagy in immune and cancer cells through increasing autophagosome formation and maturation (138). TGF-β is another factor that interacts with autophagy. Autophagy suppresses the increased TGF-β levels by reducing its degradation (139). On the other hand, autophagy stimulation in cancer cells depends on TGF-β signaling (140). In breast cancer and hepatocellular carcinoma, TGF-β has been shown to increase levels of Beclin-1, ATG5, and ATG7, thereby inducing autophagy. Then, autophagy enhances the levels of Bim and Bmf, pro-apoptotic factors, underscoring the interaction between autophagy and apoptosis (141, 142). TNF-α is another factor that regulates apoptosis and necrosis in cells and, by impairing lysosomal acidification, can suppress autophagy (143). Upon TNF-α-induced suppression of autophagy, cell death increases through oxidative damage (144). Therefore, these studies highlight that autophagy interacts with cytokines and other inflammatory factors, underscoring its complexity.
7.2 Autophagy and tumor microenvironment remodeling
7.2.1 Tumor-associated macrophages
The most abundant cell type in the TME is TAMs. (145) The macrophages resident in tissues and the recruited monocytes in the TME can differentiate into TAMs under specific conditions, driven by growth factors and chemokines secreted by cancer cells and tumor-stromal cells (146). TAMs demonstrate two types of polarization, including M1 and M2 polarization, exerting anti-cancer and tumor-promoting functions, respectively (147-149). In recent years, the role of TAMs and their polarization in regulating tumorigenesis has become increasingly important. Switching macrophage polarization and enhancing M1 polarization can improve anti-cancer immune responses (150). CAP2 can promote metastasis of gastric tumor cells by facilitating interactions between tumor cells and TAMs (151). Furthermore, TAMs have been linked to angiogenesis (152). Notably, reprogramming TAMs by downregulating Syk can suppress gemcitabine-mediated immunosuppression in pancreatic cancer (153). On the other hand, increasing TAM phagocytosis activity by enhancing cholesterol efflux can promote anti-cancer immunity in glioblastoma (154). Therefore, the function of TAMs in cancer is versatile (155-159), and this section focuses on the role of autophagy in regulating TAMs.
The IFN-γ-IDO1-kynurenine pathway-induced autophagy in cervical cancer cells enhances macrophage phagocytosis (160). IDO1 expression induced by IFN-γ led to increased autophagy in HeLa and SiHa cells, enhanced phagocytosis in these cells, and increased CD80 and CD86 expression in U937 cells. This led to autophagy in the tumor cells. Overexpression of IFN-γ and IDO1 led to depletion of tryptophan and accumulation of kynurenine in cervical cancer cells. Moreover, elevated IDO1 levels suppressed tumor growth and enhanced macrophage phagocytosis in mice. IFN-γ can trigger autophagy and phagocytosis in cervical cancer cells via IDO1 and kynurenine metabolism (160). Moreover, autophagy-driven non-traditional release of HMGB1 in glioblastoma enhances temozolomide sensitivity by inducing macrophage polarization towards an M1 phenotype (161). This suggests that reprogramming of TAMs through autophagy control can affect drug sensitivity in human cancers. The exocytosis of HMGB1 in glioblastoma cells, triggered by temozolomide, relies on secretory autophagy, leading to polarization of TAMs toward an M1-like phenotype and enhancing glioblastoma response to temozolomide. RAGE acts as the HMGB1 receptor in TAMs, triggering the RAGE-NFκB-NLRP3 inflammasome pathway to promote M1-like TAM polarization. Elevated HMGB1 levels in blood may indicate a positive response to temozolomide treatment in patients with glioblastoma. Increased autophagy secretion in glioblastoma promotes polarization of M1-like tumor-associated macrophages, enhancing the effectiveness of temozolomide.
HMGB1 is essential for communication between glioblastoma and TAMs, potentially enhancing the efficacy of temozolomide therapy (161). CD5L stimulates M2 macrophage polarization by increasing ID3 levels via autophagy (162). CD5L led cells toward a polarization comparable to that induced by IL-10. Furthermore, macrophages treated with IL-10 and CD5L exhibited higher LC3-II levels and were localized to acidic compartments, suggesting enhanced autophagy. Therefore, knocking down ATG7 with siRNA in THP1 cells inhibited CD5L-induced expression of CD163 and Mer tyrosine kinase mRNA, as well as efferocytosis. In these cells, CD5L increased ID3 transcription factor expression via ATG7. Silencing ID3 reversed the polarization caused by CD5L (162).
Blocking autophagy in breast cancer cells increases MIF expression and polarizes macrophages toward an M1 phenotype via ROS (163). MIF mRNA expression was analyzed in breast cancer tumors of different subtypes and was found to be high in the Luminal B, HER2, and Basal subtypes. MIF expression did not affect prognosis across subtypes, but inhibiting autophagy in breast cancer increased intracellular ROS levels, thereby increasing MIF expression and secretion. Secreted MIF activated MIF-regulated pathways and increased Akt and ERK phosphorylation in different cell lines. Breast cancer cells had higher levels of CD74 and CXCR2, while CXCR7 was elevated in the metastatic 4T1 cell line.
Additionally, MIF from autophagy-deficient cells induced M1 macrophage polarization (163). In addition, autophagy inhibits isoprenaline-induced M2 macrophage polarization through the ROS/ERK and mTOR axes (164). Autophagy levels decreased in macrophages exposed to tumor cell supernatant; M2 polarization was inhibited by an autophagy inducer and promoted by an autophagy inhibitor. Autophagy also reduced ROS production and M2 molecule expression. In vivo, stress-induced tumor growth and metastasis were accompanied by increased M2 polarization and ROS levels, as well as elevated mTOR and ERK signaling. Treatment with rapamycin reversed these effects. Overall, autophagy suppresses M2 macrophage polarization via the ROS/ERK and mTOR pathways in response to stress (164). Therefore, TAM polarization plays a significant role in tumorigenesis. Notably, tumor-promoting autophagy can increase macrophage M2 polarization, thereby facilitating carcinogenesis.
7.3 Cancer-associated fibroblasts
CAFs are another cell type in the TME that can regulate tumorigenesis and therapy resistance. The CAFs have been shown to secrete several growth factors, including TGF-β, HGF, and FGF, to facilitate the proliferation and survival of cancer cells. Moreover, CAFs contribute to cytokine production, including IL-6, during tumorigenesis (165). CAFs also promote angiogenesis by increasing levels of VEGF, PDGF, and IL-8, and by recruiting endothelial cells and pericytes (166). In this section, the interaction between autophagy and CAFs in the regulation of carcinogenesis is highlighted. In this context, loss of autophagy impairs CAF activation by downregulating proline biosynthesis (167). The lack of autophagy in CAFs hinders CAF activation by blocking proline biosynthesis and collagen production. Moreover, autophagy increases proline production by regulating NADK2 via mitophagy, an enzyme involved in mitochondrial NADP(H) production. In a mouse model of pancreatic cancer implanted at the original tumor site, blocking mitophagy by targeting PRKN in the supportive tissue decreased tumor mass. Therefore, blocking CAFs' mitophagy could be a promising approach for targeting the stroma in anti-cancer treatments (167).
Furthermore, CAFs utilize NUFIP1-dependent autophagy to release nucleosides and promote pancreatic tumor growth (168). CAFs are essential for the survival of pancreatic ductal adenocarcinoma in the absence of glutamine. CAFs release nucleosides via autophagy, relying on NUFIP1, which boosts glucose consumption and fuels pancreatic cancer proliferation. Nucleosides from CAFs boost glucose uptake when glutamine is lacking, and this effect is dependent on MYC. Blocking the release of nucleosides by CAFs via NUFIP1 reduced pancreatic cancer tumor weight in a mouse model, indicating a novel metabolic pathway for tumor proliferation under challenging conditions (168). Moreover, ATM activation in response to hypoxia regulates the release of autophagy-related exosomes from CAFs, thereby enhancing cancer cell invasion (169). KU60019 inhibits activated ATM, thereby preventing autophagy and exosome release from hypoxic CAFs by blocking oxidized ATM kinase activity. Furthermore, the ATM enzyme phosphorylates ATP6V1G1, thereby impairing lysosomal function and altering autophagosome fusion. GPR64 in CAF-derived exosomes activates the NF-κB pathway in breast cancer cells, boosting MMP-9 and IL-8 levels and enhancing invasiveness (169).
CAFs control bladder cancer invasion and metabolic features via autophagy (170). Rapamycin and siRNA were used to control autophagy in cancer-related fibroblasts. Rapamycin enhanced autophagy in CAFs, thereby increasing T24 cell proliferation and invasion. Increased lactate levels were observed in both groups, with the autophagy-enhanced group exhibiting higher expression of genes associated with metabolism and invasion (170). CAFs enhance the recovery of cancer cells following radiation therapy by stimulating autophagy (171). CAFs promote cancer cell recovery and tumor recurrence after radiotherapy by inducing autophagy via the release of IGF1/2, CXCL12, and β-hydroxybutyrate. These molecules derived from CAFs increase ROS levels after irradiation, thereby enhancing PP2A activity, reducing mTOR activation, and promoting autophagy in cancer cells. Blocking IGF2 and using an autophagy inhibitor reduces tumor recurrence after radiotherapy in mice with CAFs. Focusing on the autophagy process in cancer cells may offer a promising approach to enhance the efficacy of radiotherapy (171).
Additionally, autophagy in CAFs boosts the advancement of triple-negative breast cancer cells (172). CAFs were characterized by elevated α-smooth muscle actin (α-SMA) levels. Levels of autophagy-related Beclin 1 and LC3-II/I protein conversion were significantly elevated in CAFs compared to normal fibroblasts (NFs). The breast cancer cells showed enhanced migration, invasion, and proliferation when exposed to Conditioned medium from CAFs (CAFs-CM) compared with the other three groups. In the CAFs-CM group, breast cancer cells showed increased levels of vimentin and N-cadherin proteins, while the level of E-cadherin protein decreased compared to the control group. Additional research showed increased β-catenin and P-GSK-3β protein levels, essential proteins in the Wnt/β-catenin pathway, in the CAFs-CM group compared with the control group (172).
7.4 Dendritic cells
DCs are essential APCs that regulate immune responses. The induction of DCs is vital for stimulating immune responses. The tumor antigens are captured by DCs and presented to T cells via MHC molecules. This can induce CTLs to eliminate tumor cells (173). The migration of DCs from peripheral tissues to lymph nodes to prime naïve T cells for the induction of adaptive immune responses. Moreover, they can secrete IL-12, which promotes T cell differentiation and proliferation (174). DC function can be affected by autophagy. Naringenin regulates the FKBP4/NR3C1/NRF2 axis in breast cancer autophagy and proliferation, as well as in DC differentiation and maturation (175). The FKBP4/NR3C1 axis negatively regulates NRF2 at the protein level in human breast cancer cells. FKBP4 was found to regulate NRF2 by facilitating NR3C1 nuclear translocation. Naringenin, a flavonoid found in citrus and tomatoes, can inhibit autophagy and cell growth in breast cancer cells by modulating the FKBP4/NR3C1/NRF2 pathway. Naringenin also enhanced dendritic cell differentiation and maturation via the same pathway. This research proposes that targeting the FKBP4/NR3C1/NRF2 pathway could be a promising approach to treating breast cancer (175). Cancer cells in the lungs release exosomal MALAT1 to prevent DC phagocytosis, triggering an inflammatory response, upregulating costimulatory molecules, and promoting DC autophagy via the AKT/mTOR pathway (176). LLC and A549 cells, as well as their exosomes, exhibited higher MALAT1 expression than Beas-2b cells, with LLC cells displaying the highest levels. Suppressing MALAT1 reduced both tumor cell growth and tumor size. Inhibition also increased DC particle uptake, inflammation, and T cell growth, while decreasing DC self-degradation and T cell maturation. PEX groups showed elevated levels of p-AKT, AKT, p-mTOR, and mTOR, while siMALAT1 groups displayed decreased levels (176). Moreover, chloroquine blocks drug-induced autophagy in HCT-116 colon cancer cells, improving DC maturation and T cell responses triggered by tumor cell lysate (177). It was investigated how co-administration of chloroquine, an autophagy-inhibiting drug, with low doses of 5-fluorouracil (5-FU) affects tumor cells' ability to promote DC maturation. DCs exposed to lysates from tumor cells treated with this combination displayed increased maturation and activation. The developed DCs enhanced the activation of CD4+ and CD8+ T cells, resulting in increased proliferation and IFN-γ secretion. The cocultures also enhanced the production of cytotoxic T cells. They increased the expression of genes that inhibit tumor growth and decreased the expression of genes associated with cancer progression and metastasis. On the whole, the pairing of 5-FU and CQ increased the maturation of tumor cell-induced DCs and boosted the T cell responses mediated by DCs (177).
In addition, effective cancer immunotherapy is enabled by manipulating DCs in their natural environment with a nanoactivator that regulates autophagy (178). A nanoactivator was designed to increase autophagy in vivo, thereby enhancing antigen presentation by DCs and generating antigen-specific T cells. The nanoactivator significantly enhances the presentation of tumor antigens and T-cell priming. The nanoactivators effectively reduce tumor proliferation and extend mouse lifespan. These findings suggest that manipulating in situ DCs via induction of autophagy is a promising approach to enhance antigen presentation and eliminate tumors (178). Lactosylated N-Alkyl polyethylenimine-coated iron oxide nanoparticles induced autophagy in mouse DCs (179). Autophagy was employed as a crucial tool to investigate how DCs respond to labeled nanoparticles. The nanoparticles trigger a defensive autophagy response in DCs, as blocking autophagy can lead to cell death. At the same time, nanoparticle-induced autophagy can support DC maturation, a crucial step in their migration and antigen presentation. Autophagy-triggered DC maturation has been shown to enhance DC vaccine efficacy, suggesting that this probe could enhance therapeutic immune activation in addition to its MRI-tracking capabilities. Iron oxide nanoparticles coated with nimine triggered autophagy in mouse dendritic cells (179).
7.5 Natural killer cells
NK cells are cytotoxic lymphocytes vital to the innate immune system. NK cells are responsible for eliminating tumor cells before sensitization. NK cells can release granules containing proteins and granzymes to eradicate cancer cells, triggering apoptosis. There is no requirement for prior antigen exposure, providing an immediate response for cancer cell elimination (180). The identification of cancer cells relies on stress-mediated ligands, including MICA/B and ULBP, binding activating receptors on NK cells, such as NKG2D, to induce a cytotoxic response (181). The interaction between autophagy and NK cells is observed within the TME. Artesunate (ART)-triggered ATG5-associated autophagy enhances the killing capacity of NK92 cells against endometrial cancer cells via CD155 and CD226/TIGIT interactions (182). ART tility inhibits the growth and movement of UCEC cells and induces cell death and autophagy. ART-induced autophagy increased CD155 levels in UCEC cells, boosting NK92 cell cytotoxicity by altering CD155 interactions with its receptors. ART regulated CD155, with ATG5 contributing to some extent; decreasing ATG5 levels reduced both CD155 expression and NK92 cell cytotoxicity (182). However, further investigation of the NK-autophagy interaction is required.
7.6 T lymphocytes
The majority of triple-negative breast cancer patients do not exhibit a response to T cell-based immunotherapies. Regrettably, there remains a lack of understanding of the molecular determinants. A lack of autophagy is genetically associated with breast cancer. Impaired autophagy in TNBC cells hinders T cell-induced tumor destruction in both laboratory and living systems (183). A lack of autophagy results in immune suppression via Skp2-mediated Lys63 ubiquitination of Tenascin-C at specific sites. Elevated levels of Tenascin-C are associated with a poor prognosis and a weakened immune response. Blocking Tenascin-C in the context of impaired autophagy increases T cell-mediated tumor destruction and improves the efficacy of anti-PD1/PDL1 treatment, suggesting potential as a successful therapeutic strategy (183). Autophagy signaling is necessary for CAR-T cells to effectively eliminate B-cell malignancies, suggesting that targeting autophagy could enhance CAR-T cell therapy (184). CRISPR/Cas9 knockout screening revealed that overexpression of autophagy genes (ATG3, BECN1, RB1CC1) helped shield cancer cells from CD19 CAR-T cell-induced cell death. Studies conducted outside living organisms have corroborated this finding, linking increased levels of autophagy-related proteins in tumors to reduced patient response and survival after treatment. Autophagy is involved in treatment responses and recurrence after CD19 CAR-T therapy. In mouse models, tumor cell sensitivity to CD19 CAR-T killing was heightened by inhibiting autophagy and knocking out RB1CC1. Cancer-induced autophagy prevents CAR-T cell attack by activating the TNF-α-TNFR1 axis to induce apoptosis and by activating STAT1/IRF1-mediated chemokine signaling (184). The impact of fasting-mimicking diets (FMDs) on hematologic malignancies when combined with standard therapies remains poorly understood, despite their effectiveness in treating various solid tumors in mouse models. The fasting-mimicking diet suppresses autophagy and, when combined with chemotherapy, enhances T-cell-mediated survival without leukemia (185). In rodents, alternating between a diet mimicking fasting and vincristine treatment increased cancer-free survival compared with a standard diet alone. Vincristine decreased levels of autophagy markers, and fasting further reduced them. Chloroquine may serve as a replacement for fasting and enhance survival when combined with vincristine. Blocking autophagy genes ULK1 and ATG9a increased vincristine's toxicity. Anti-CD8 antibodies counteracted the effects of vincristine and fasting, suggesting that the immune system contributes to leukemia-free survival. Factors contributing to mice surviving cancer-free while fasting include inhibition of autophagy and strengthening of the immune response (185).
Moreover, autophagy shields tumors from T cell-induced cell death by blocking TNFα-triggered apoptosis (186). A mouse genome-wide CRISPR knockout screen was performed to assess the role of TNFα signaling in tumor cells in T cell-induced apoptosis. NF-κB signaling and autophagy were identified as crucial protective mechanisms. Inhibiting autophagy genes rendered tumor cells more susceptible to T-cell-mediated destruction. Blocking mTOR led to increased autophagy, which, in turn, protected tumor cells. Autophagy functions early in the TNFα pathway to restrict caspase-8 activation. Enhancing T cell-based immunotherapies may be achieved by blocking tumor cell autophagy, thereby improving the effectiveness of immune checkpoint blockade (186). In addition, the temporary inhibition of autophagy permanently impairs metabolic activity in lung tumor cells and enhances T-cell-induced tumor destruction (187). A transient reduction in ATG5 expression throughout the body results in reduced lung tumor growth. This interference reduces glucose and lactate uptake in tumors, thereby affecting their metabolism and growth. Surprisingly, T cell penetration into tumors was enhanced in the absence of ATG5, facilitating tumor eradication. Despite the temporary restoration of autophagy, tumor metabolism remains changed, and T cell infiltration remains increased.
Furthermore, periodic suppression of ATG5 increased the longevity of mice with lung tumors and holds potential for upcoming cancer treatments (187). In addition, inhibition of FIP200 and autophagy by tumor-produced lactate promotes apoptotic cell death in naive T cells and impairs tumor immunity (188). FIP200 depletion induces programmed cell death in untreated T cells from individuals with ovarian cancer and in tumors from mice, thereby impairing the body's ability to fight cancer by disrupting autophagy, promoting mitochondrial dysfunction, and elevating ROS levels. In terms of mechanisms, FIP200 depletion disrupts the balance between pro- and anti-apoptotic factors in T cells by increasing Ago2 degradation, reducing Ago2-microRNA1198-5p complex formation, and inhibiting microRNA1198-5p biogenesis. Tumors can evade the immune response by targeting T cells metabolically and suppressing FIP200 expression via lactate produced by the tumor. Increasing Bcl-2 levels and blocking mitochondrial complex I could prevent T cell death and bolster the body's ability to fight tumors (188).
7.7 Treg cells
Treg cells are a subset of T cells that help maintain immune tolerance and reduce autoimmunity. Treg cells can promote immune evasion and carcinogenesis. Treg cells suppress T-cell activation through the production of IL-10 and TGF-β, thereby reducing the immune response and facilitating immune evasion (189). The presence and accumulation of Treg cells in the TME can affect other cells, including immune cells, stromal cells, and cancer cells, thereby mediating immunosuppression and supporting tumor growth and metastasis (190). Another function of Treg cells in reducing immune surveillance is their inhibition of DC maturation and function, thereby decreasing T cell activation (191). In recent years, the role of Treg cells in regulating immunosuppression has become increasingly important (192-194). This section evaluates the regulation of Treg cells by autophagy in human cancers. CD39, an ectonucleotidase expressed on Treg cells in humans, plays a vital role in immune regulation; however, its function is disrupted in autoimmune conditions and cancer-related immune suppression. The expression of CD39 on Treg cells is not influenced by the transcription factors FOXP3 and HELIOS but is instead enhanced by the canonical TGF-β and mTOR signaling pathways. Moreover, the TGF-β-induced increase in CD39 was counteracted by ROS-driven autophagy (195). CD39+ peripheral blood Treg cells are a distinct subset characterized by minimal autophagic flux and no ROS generation. High levels of CD39+ Treg cells are observed in patients with rare genetic defects that affect autophagy. The specific transcriptional program guides these processes by reducing NEFL and PLAC8 expression in CD39+ Treg cells and increasing SOX4 levels. SOX4 overexpression increases CD39 levels in Treg cells. In contrast, its deletion leads to the opposite effect, highlighting the crucial role of SOX4 in regulating the immune system and in the crosstalk between tolerogenic signals and autophagy in Treg cells (195).
7.8 Myeloid-derived suppressor cells
MDSCs are a heterogeneous population of immature myeloid cells that expand during tumorigenesis and other pathological events. They can also mediate immunosuppression. The immunosuppressive activity of MDSCs is mediated by ARG1, ROS, and iNOS, which suppress T cell activation and proliferation and deplete nutrients, including arginine and cysteine, and impair T cell function (196). The secretion of chemokines and cytokines by MDSCs within the TME can exert immunosuppressive effects by recruiting Treg cells. Moreover, MDSCs can reprogram TAMs into a tumorigenic phenotype (197). The underlying mechanisms of MDSC immunosuppression have been highlighted (198-202), and this section focuses on the interaction between autophagy and MDSCs in human cancers.
In this context, autophagy controls the accumulation and function of granulocytic MDSCs via STAT3 in endotoxin shock (203). There was an increase in granulocytic MDSCs with heightened autophagy activity in mice exposed to lipopolysaccharide (LPS), whereas monocytic MDSCs (M-MDSCs) did not demonstrate this accumulation. Boosting ROS production enhanced the immunosuppressive activity of granulocytic MDSC during M1 macrophage polarization by increasing their accumulation through inhibition of autophagy. This hindrance also enhanced STAT3 phosphorylation in G-MDSCs, which is essential for their gathering and activity. Pharmacological inhibition of autophagy exacerbated the condition of LPS-challenged mice in vivo. The results indicate that autophagy plays a critical role in regulating MDSC accumulation and function via STAT3 signaling, providing insight into the regulation of the immune response and the development of inflammatory diseases (203). The nanoassembly, responsive to redox conditions, inhibited MDSC recruitment by silencing lactate dehydrogenase A via autophagy, thereby improving the efficacy of cancer immunochemotherapy (204). Three GSH-responsive polymers form a nanoassembly that responds to redox reactions, using PVL and DA. It contains doxorubicin. Cellular uptake and tumor-targeted delivery are enhanced by integrin binding to the RGD ligand. Following GSH-induced cleavage of DA, there was a rapid release of drugs and inhibition of LDHA, resulting in decreased cytokine production, reduced MDSC recruitment, and improved anti-tumor immune responses. This treatment demonstrates potent anticancer activity against 4T1 tumors, opening new opportunities for immunochemotherapy (204). Moreover, autophagy shows a positive correlation with MDSC accumulation in 4-nitroquinoline-1-oxide-induced oral cancer (205). There was an increase in the presence of autophagy markers such as LC3B, p62, and Beclin 1 in both 4NQO-induced carcinogenesis and oral cancer in humans. During oral cancer development, the numbers of MDSCs and Tregs also increased. Moreover, there was a significant correlation between LC3B and p62 levels and the presence of MDSCs, whereas Beclin-1 expression correlated with the rise in Tregs (205).
Autophagy coordinates the regulatory mechanisms of tumor-associated MDSCs (206). Elevated levels of functional autophagy were observed in MDSCs from individuals with melanoma and in mouse models of melanoma. Disruption of autophagy in myeloid cells slowed tumor progression and enhanced antitumor immune responses. M-MDSCs lacking autophagy had reduced suppressive function and higher levels of MHC class II molecules on their surface, thereby activating tumor-specific CD4+ T cells. Inhibiting the MARCH 1 E3 ligase, which controls MHC II degradation in M-MDSCs, reduced tumor size and enhanced the body's ability to fight cancer (206).
Additionally, the survival of early-stage MDSCs in breast cancer is supported by the activation of the Wnt/mTOR pathway due to the repression of autophagy caused by the lack of SOCS3 (207). MDSCs in the early stages of SOCS3MyeKO mice showed blocked differentiation in the myeloid lineage due to restricted autophagy activation, in a Wnt/mTOR-dependent manner. The downregulation of C/EBPβ by miR-155 had activated the Wnt/mTOR pathway, leading to the inhibition of autophagy and arrest of differentiation in early-stage MDSCs. Moreover, blocking Wnt/mTOR signaling inhibited tumor growth and the immunosuppressive activities of early-stage MDSCs (207). HMGB1 enhances MDSC survival by triggering autophagy (208). Blocking autophagy or HMGB1 promotes apoptotic MDSCs and reduces autophagy, thereby supporting their survival. MDSCs in circulation exhibit autophagy, whereas tumor-infiltrating cells show even greater autophagy, which promotes tumor progression. Inflamed, low-oxygen environments in solid tumors promote the accumulation of immunosuppressive MDSCs. Autophagy prolongs the survival and viability of myeloid-derived suppressor cells (208).
7.9 Neutrophils
As blood cells, neutrophils participate in the innate immune system. Despite neutrophils' role in infections, recent studies have highlighted their role in tumorigenesis. Notably, neutrophils can secrete inflammatory cytokines, chemokines, and ROS to mediate tumor-promoting inflammation. These factors can enhance the proliferation and survival of cancer cells (209). Moreover, neutrophils can release MMPs and elastases that degrade the extracellular matrix, thereby enhancing the invasion and metastasis of tumor cells (210). In recent years, the role of neutrophils in regulating immunosuppression has become increasingly important (211-214). Moreover, there is a correlation between autophagy and neutrophils in human cancers. The enhanced autophagy supports the survival and pro-tumor effects of neutrophils in human liver cancer (215). The factors secreted by hepatoma cells induce high LC3 expression in neutrophils within hepatocellular carcinoma. Blocking Erk1/2, p38, and NF-κB pathways can decrease tumor-triggered autophagy in these neutrophils. Autophagy in neutrophils results in persistent effects, elevated production of oncostatin M and MMP-9, and enhanced cancer cell migration. This autophagic process is not associated with inhibition of mTOR signaling and is essential for cell viability (215). Figure 4 highlights the association between macrophages and TME components.
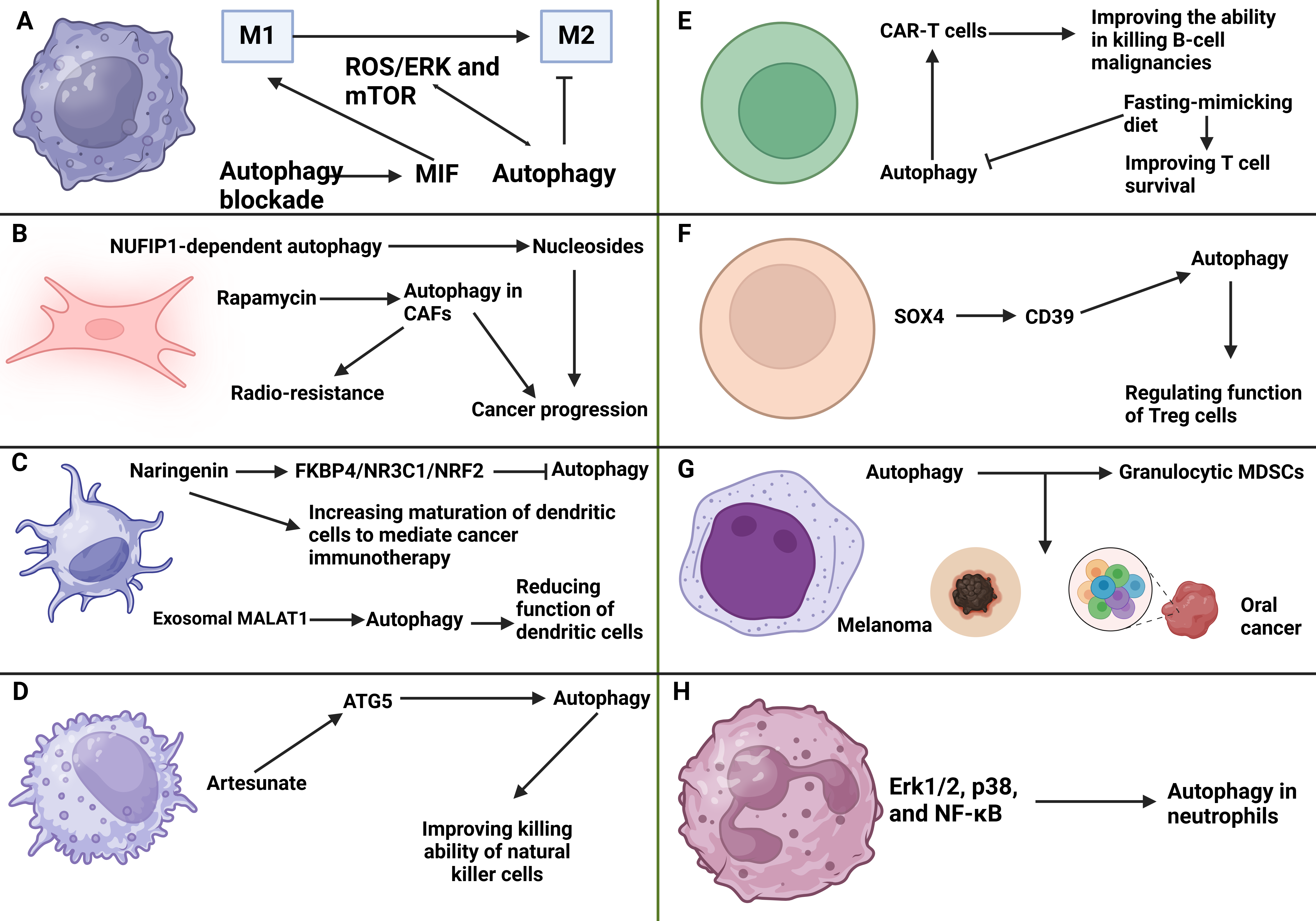
Figure 4: The interaction of autophagy and tumor microenvironment components. A) The inhibition of autophagy in macrophages can increase MIF levels to promote M1 polarization. Moreover, autophagy demonstrates interaction with mTOR and ROS/ERK axis to suppress M2 polarization of macrophages; B) The NUFIP1-related autophagy can secrete nucleosides to promote cancer progression. Moreover, autophagy induction of cancer-associated fibroblasts can mediate radio-resistance; C) The suppression of autophagy by naringenin can promote maturation of dendritic cells in cancer immunotherapy. Exosomal MALAT1 induces autophagy to impair the function of dendritic cells; D) On the other hand, artesunate can increase ATG5 levels to induce autophagy in accelerating the function of natural killer cells; E) The function of CAR-T cells in the elimination of tumor cells is improved by autophagy induction. Autophagy inhibition by fast-mimicking diet can promote survival of T cells; F) The regulation of autophagy by the SOX4/CD39 axis is vital for affecting the function of Treg cells; G) The induction of autophagy can promote levels of granulocytic MDSCs in promoting progression of melanoma and oral cancer; H) ERK1/2- p38 and NF-κB stimulate autophagy in neutrophils in cancer progression. This figure was created using BioRender.
Discussion
This review highlights the potential of autophagy in regulating cancer immunotherapy and TME remodeling. In the immune system, autophagy has been associated with the regulation of innate and adaptive immune responses. Notably, autophagy can regulate the immune evasion in human cancers. One example is the role of autophagy in degrading MHC molecules, thereby facilitating immune evasion. Therefore, the therapeutic regulation of autophagy is essential for overcoming immune resistance. Notably, autophagy has the potential to regulate ICD. The pro-death autophagy facilitates ICD to promote maturation of DCs for the induction of T cells in cancer immunotherapy. Notably, changes in autophagy in response to hypoxia, chemokines, and ILs can also be linked to the immune system. In addition, autophagy has been suggested to be a critical regulator of the TME in human cancers. TME has been comprised of TAMs, neutrophils, CAFs, Treg cells, MDSCs, and other cell types. Since TME components can regulate immune responses, and autophagy can modulate the TME, therapeutic regulation of autophagy may open new avenues for treating human cancers by facilitating immune responses. For example, pro-survival autophagy in TAMs can drive M2 polarization, thereby enhancing tumorigenesis and creating an immunosuppressive TME. Moreover, autophagy is vital for CAF activation. The presence of autophagy and its interaction with various TME components, such as Treg cells, can suppress the function of DCs and T cells in the immune system. As a result, regulating autophagy is vital for remodeling the TME to potentiate cancer immunotherapy.
References
1. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591‑5596. https://doi.org/10.1242/jcs.116392
2. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541 550. https://doi.org/10.1038/s41591-018-0014-x
3. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014-1022. https://doi.org/10.1038/ni.2703
4. Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Al Mazeedi MAM, et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int J Mol Sci. 2017;18(7):1586. https://doi.org/10.3390/ijms18071586
5. Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T. Regulation of tumor growth and metastasis: The role of tumor microenvironment. Cancer Growth Metastasis. 2014;7:9-18. https://doi.org/10.4137/cgm.s11285
6. Xia S, Lal B, Tung B, Wang S, Goodwin CR, Laterra J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016;18(4):507-517. https://doi.org/10.1093/neuonc/nov171
7. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174-186. https://doi.org/10.1038/s41568-019-0238-1
8. Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. 2021;18(12):792-804. https://doi.org/10.1038/s41571-021-00546-5
9. Ping Q, Yan R, Cheng X, Wang W, Zhong Y, Hou Z, et al. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021;28(9):984-999. https://doi.org/10.1038/s41417-021-00318-4
10. Xing F, Saidou J, Watabe K. Cancer-associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 2010;15(1):166-179. https://doi.org/10.2741/3613
11. Li X, Liu R, Su X, Pan Y, Han X, Shao C, Shi Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18(1):177. https://doi.org/10.1186/s12943-019-1102-3
12. Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity. 2014;41(1):49-61. https://doi.org/10.1016/j.immuni.2014.06.010
13. Wang Y, Wang D, Yang L, Zhang Y. Metabolic reprogramming in the immunosuppression of tumor-associated macrophages. Chin Med J (Engl). 2022;135(20):2405-2416. https://doi.org/10.1097/cm9.0000000000002426
14. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69 Suppl 3:4-10. https://doi.org/10.1159/000088478
15. Liao D, Johnson RS. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26(2):281-290. https://doi.org/10.1007/s10555-007-9066-y
16. Goudar RK, Vlahovic G. Hypoxia, angiogenesis, and lung cancer. Curr Oncol Rep. 2008;10(4):277-282. https://doi.org/10.1007/s11912-008-0043-6
17. Kleiner DE, Stetler-Stevenson WG. Matrix metalloproteinases and metastasis. Cancer Chemother Pharmacol. 1999;43 Suppl:S42-51. https://doi.org/10.1007/s002800051097
18. Foda HD, Zucker S. Matrix metalloproteinases in cancer invasion, metastasis and angiogenesis. Drug Discov Today. 2001;6(9):478-482. https://doi.org/10.1016/s1359-6446(01)01752-4
19. Itoh Y, Nagase H. Matrix metalloproteinases in cancer. Essays Biochem. 2002;38:21-36. https://doi.org/10.1042/bse0380021
20. Dzobo K, Senthebane DA, Dandara C. The tumor microenvironment in tumorigenesis and therapy resistance revisited. Cancers (Basel). 2023;15(2):376. https://doi.org/10.3390/cancers15020376
21. Lu Q, Kou D, Lou S, Ashrafizadeh M, Aref AR, Canadas I, et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J Hematol Oncol. 2024;17(1):16. https://doi.org/10.1186/s13045-024-01535-8
22. Elmusrati A, Wang J, Wang CY. Tumor microenvironment and immune evasion in head and neck squamous cell carcinoma. Int J Oral Sci. 2021;13(1):24. https://doi.org/10.1038/s41368-021-00131-7
23. Kim SK, Cho SW. The evasion mechanisms of cancer immunity and drug intervention in the tumor microenvironment. Front Pharmacol. 2022;13:868695. https://doi.org/10.3389/fphar.2022.868695
24. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. 2021;12:636568. https://doi.org/10.3389/fimmu.2021.636568
25. Garrido F, Aptsiauri N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology. 2019;158(4):255-266. https://doi.org/10.1111/imm.13114
26. Garcia-Lora A, Algarra I, Garrido F. MHC class I antigens, immune surveillance, and tumor immune escape. J Cell Physiol. 2003;195(3):346-355. https://doi.org/10.1002/jcp.10290
27. Ye Y, Xu Y, Lai Y, He W, Li Y, Wang R, et al. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J Cell Biochem. 2018;119(3):2951-2963. https://doi.org/10.1002/jcb.26509
28. Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest. 2007;117(5):1167-1174. https://doi.org/10.1172/jci31202
29. De Cicco P, Ercolano G, Ianaro A. The new era of cancer immunotherapy: Targeting myeloid-derived suppressor cells to overcome immune evasion. Front Immunol. 2020;11:1680. https://doi.org/10.3389/fimmu.2020.01680
30. Wang L, Liu JQ, Talebian F, El-Omrani HY, Khattabi M, Yu L, et al. Tumor expression of CD200 inhibits IL-10 production by tumor-associated myeloid cells and prevents tumor immune evasion of CTL therapy. Eur J Immunol. 2010;40(9):2569-2579. https://doi.org/10.1002/eji.201040472
31. Beck C, Schreiber H, Rowley D. Role of TGF-beta in immune-evasion of cancer. Microsc Res Tech. 2001;52(4):387-395. https://doi.org/10.1002/1097-0029(20010215)52:4<387::aid-jemt1023>3.0.co;2-w
32. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. 2018;6(1):57. https://doi.org/10.1186/s40425-018-0360-8
33. Zamanakou, M., A.E. Germenis, and V. Karanikas, Tumor immune escape mediated by indoleamine 2, 3-dioxygenase. Immunology letters, 2007. 111(2): p. 69-75. https://doi.org/10.1016/j.imlet.2007.06.001
34. Labi V, Erlacher M. How cell death shapes cancer. Cell Death Dis. 2015;6(3):e1675. https://doi.org/10.1038/cddis.2015.20
35. Chen X, Zeh HJ, Kang R, Kroemer G, Tang D. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol. 2021;18(11):804-823. https://doi.org/10.1038/s41575-021-00486-6
36. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860-875. https://doi.org/10.1038/nrc3380
37. Li X, He S, Ma B. Autophagy and autophagy-related proteins in cancer. Mol Cancer. 2020;19(1):12. https://doi.org/10.1186/s12943-020-1138-4
38. Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24(8):560-575. https://doi.org/10.1038/s41580-023-00585-z
39. Mulcahy Levy JM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528-542. https://doi.org/10.1038/nrc.2017.53
40. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726-734. https://doi.org/10.1038/nrc1692
41. Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun. 2018;9(1):1944. https://doi.org/10.1038/s41467-018-04070-6
42. Le Bot N. Autophagy: a new regulator of development. Nat Cell Biol. 2007;9(7):741. https://doi.org/10.1038/ncb0707-741
43. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131-1135. https://doi.org/10.1038/nature07976
44. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466(7302):68-76. https://doi.org/10.1038/nature09204
45. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323-335. https://doi.org/10.1038/nature09782
46. Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177(7):1682-1699. https://doi.org/10.1016/j.cell.2019.05.026
47. Qin Y, Ashrafizadeh M, Mongiardini V, Grimaldi B, Crea F, Rietdorf K, et al. Autophagy and cancer drug resistance in dialogue: Pre-clinical and clinical evidence. Cancer Lett. 2023;570:216307. https://doi.org/10.1016/j.canlet.2023.216307
48. Ashrafizadeh M, Zhang W, Zou R, Sethi G, Klionsky DJ, Zhang X. A bioinformatics analysis, pre-clinical and clinical conception of autophagy in pancreatic cancer: Complexity and simplicity in crosstalk. Pharmacol Res. 2023;194:106822. https://doi.org/10.1016/j.phrs.2023.106822
49. Yang Y, Liu L, Tian Y, Gu M, Wang Y, Ashrafizadeh M, et al. Autophagy-driven regulation of cisplatin response in human cancers: Exploring molecular and cell death dynamics. Cancer Lett. 2024;587:216659. https://doi.org/10.1016/j.canlet.2024.216659
50. Zheng Y, Zhan Y, Zhang Y, Zhang Y, Liu Y, Xie Y, et al. Hexokinase 2 confers radio-resistance in hepatocellular carcinoma by promoting autophagy-dependent degradation of AIMP2. Cell Death Dis. 2023;14(8):488. https://doi.org/10.1038/s41419-023-06009-2
51. Ferretti GDS, Quaas CE, Bertolini I, Zuccotti A, Saatci O, Kashatus JA, et al. HSP70-mediated mitochondrial dynamics and autophagy represent a novel vulnerability in pancreatic cancer. Cell Death Differ. 2024;31(7):881-896. https://doi.org/10.1038/s41418-024-01310-9
52. Liang Y, Chen B, Xu F, Long L, Ye F, Wang Y, et al. LncRNA PRBC induces autophagy to promote breast cancer progression through modulating PABPC1-mediated mRNA stabilization. Oncogene. 2024;43(14):1019-1032. https://doi.org/10.1038/s41388-024-02971-z
53. Hoenigsperger H, Koepke L, Acharya D, Hunszinger V, Freisem D, Grenzner A, et al. CSNK2 suppresses autophagy by activating FLN-NHL-containing TRIM proteins. Autophagy. 2024;20(5):994-1014. https://doi.org/10.1080/15548627.2023.2281128
54. Mukhopadhyay S, Encarnacion-Rosado J, Kimmelman AC. Autophagy fuels mitochondrial function through regulation of iron metabolism in pancreatic cancer. Autophagy. 2024;20(4):963-964. https://doi.org/10.1080/15548627.2023.2223473
55. Huang P, Zhao H, Pan X, Li J, Pan W, Dai H, et al. SIRT3-mediated autophagy contributes to ferroptosis-induced anticancer by inducing the formation of BECN1-SLC7A11 complex. Biochem Pharmacol. 2023;213:115592. https://doi.org/10.1016/j.bcp.2023.115592
56. Feng T, Tang Z, Karges J, Shen J, Jin C, Chen Y, et al. Exosome camouflaged coordination-assembled Iridium(III) photosensitizers for apoptosis-autophagy-ferroptosis induced combination therapy against melanoma. Biomaterials. 2023;301:122212. https://doi.org/10.1016/j.biomaterials.2023.122212
57. Xiao C, Sun Y, Fan J, Nguyen W, Chen S, Long Y, et al. Engineering cannabidiol synergistic carbon monoxide nanocomplexes to enhance cancer therapy via excessive autophagy. Acta Pharm Sin B. 2023;13(11):4591-4606. https://doi.org/10.1016/j.apsb.2023.05.019
58. Yang X, Liu Z, Xu X, He M, Xiong H, Liu L. Casticin induces apoptosis and cytoprotective autophagy while inhibiting stemness involving Akt/mTOR and JAK2/STAT3 pathways in glioblastoma. Phytother Res. 2024;38(1):305-320. https://doi.org/10.1002/ptr.8048
59. Deng Z, Shen D, Yu M, Zhou F, Shan D, Fang Y, et al. Pectolinarigenin inhibits bladder urothelial carcinoma cell proliferation by regulating DNA damage/autophagy pathways. Cell Death Discov. 2023;9(1):214. https://doi.org/10.1038/s41420-023-01508-9
60. Chen Z, Chen H, Huang L, Duan B, Dai S, Cai W, et al. ATB0,+-targeted nanoparticles initiate autophagy suppression to overcome chemoresistance for enhanced colorectal cancer therapy. Int J Pharm. 2023;641:123082. https://doi.org/10.1016/j.ijpharm.2023.123082
61. Wang L, Zhu H, Shi Z, Chen B, Huang H, Lin G, et al. MK8722 initiates early-stage autophagy while inhibiting late-stage autophagy via FASN-dependent reprogramming of lipid metabolism. Theranostics. 2024;14(1):75-95. https://doi.org/10.7150/thno.83051
62. Gu C, Liu X, Luo L, Chen J, Zhou X, Chen G, et al. Metal-DNA nanocomplexes enhance chemo-dynamic therapy by inhibiting autophagy-mediated resistance. Angew Chem Int Ed Engl. 2023;62(50):e202307020. https://doi.org/10.1002/anie.202307020
63. Zhang X, Lao M, Yang H, Sun K, Dong Y, He L, et al. Targeting cancer-associated fibroblast autophagy renders pancreatic cancer eradicable with immunochemotherapy by inhibiting adaptive immune resistance. Autophagy. 2024;20(6):1314-1334. https://doi.org/10.1080/15548627.2023.2300913
64. Jiang GM, Tan Y, Wang H, Peng L, Chen HT, Meng XJ, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18(1):17. https://doi.org/10.1186/s12943-019-0944-z
65. Pan H, Chen L, Xu Y, Han W, Lou F, Fei W, et al. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget. 2016;7(16):21235-46. https://doi.org/10.18632/oncotarget.6908
66. Fang L, Wu HM, Ding PS, Liu RY. TLR2 mediates phagocytosis and autophagy through JNK signaling pathway in Staphylococcus aureus-stimulated RAW264.7 cells. Cell Signal. 2014;26(4):806-14. https://doi.org/10.1016/j.cellsig.2013.12.016
67. Lu Z, Xie D, Chen Y, Tian E, Muhammad I, Chen X, et al. TLR2 mediates autophagy through ERK signaling pathway in Mycoplasma gallisepticum-infected RAW264.7 cells. Mol Immunol. 2017;87:161-170. https://doi.org/10.1016/j.molimm.2017.04.013
68. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27(7):1110-21. https://doi.org/10.1038/emboj.2008.31
69. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135-44. https://doi.org/10.1016/j.immuni.2007.05.022
70. Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H, et al. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10(2):257-68. https://doi.org/10.4161/auto.27162
71. van der Vaart M, Korbee CJ, Lamers GE, Tengeler AC, Hosseini R, Haks MC, et al. The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense (corrected). Cell Host Microbe. 2014;15(6):753-67. https://doi.org/10.1016/j.chom.2014.05.005
72. Lupfer C, Kanneganti TD. The expanding role of NLRs in antiviral immunity. Immunol Rev. 2013;255(1):13-24. https://doi.org/10.1111/imr.12089
73. Baker K, Rath T, Lencer WI, Fiebiger E, Blumberg RS. Cross-presentation of IgG-containing immune complexes. Cell Mol Life Sci. 2013;70(8):1319-34. https://doi.org/10.1007/s00018-012-1100-8
74. Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008;68(17):6889-95. https://doi.org/10.1158/0008-5472.can-08-0161
75. Fiegl D, Kägebein D, Liebler-Tenorio EM, Weisser T, Sens M, Gutjahr M, et al. Amphisomal route of MHC class I cross-presentation in bacteria-infected dendritic cells. J Immunol. 2013;190(6):2791-806. https://doi.org/10.4049/jimmunol.1202741
76. Bell C, English L, Boulais J, Chemali M, Caron-Lizotte O, Desjardins M, et al. Quantitative proteomics reveals the induction of mitophagy in tumor necrosis factor-α-activated (TNFα) macrophages. Mol Cell Proteomics. 2013;12(9):2394-407. https://doi.org/10.1074/mcp.m112.025775
77. Li B, Lei Z, Lichty BD, Li D, Zhang GM, Feng ZH, et al. Autophagy facilitates major histocompatibility complex class I expression induced by IFN-γ in B16 melanoma cells. Cancer Immunol Immunother. 2010;59(2):313-21. https://doi.org/10.1007/s00262-009-0752-1
78. Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011;186(3):1564-74. https://doi.org/10.4049/jimmunol.1001822
79. Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182(7):4046-55. https://doi.org/10.4049/jimmunol.0801143
80. Parekh VV, Wu L, Boyd KL, Williams JA, Gaddy JA, Olivares-Villagómez D, et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J Immunol. 2013;190(10):5086-101. https://doi.org/10.4049/jimmunol.1202071
81. Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A. 2012;109(22):8670-5. https://doi.org/10.1073/pnas.1205305109
82. Kumai T, Matsuda Y, Ohkuri T, Oikawa K, Ishibashi K, Aoki N, et al. c-Met is a novel tumor associated antigen for T-cell based immunotherapy against NK/T cell lymphoma. Oncoimmunology. 2015;4(2):e976077. https://doi.org/10.4161/2162402x.2014.976077
83. Clarke AJ, Ellinghaus U, Cortini A, Stranks A, Simon AK, Botto M, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74(5):912-20. https://doi.org/10.1136/annrheumdis-2013-204343
84. Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4(3):309-14. https://doi.org/10.4161/auto.5474
85. Conway KL, Kuballa P, Khor B, Zhang M, Shi HN, Virgin HW, et al. ATG5 regulates plasma cell differentiation. Autophagy. 2013;9(4):528-37. https://doi.org/10.4161/auto.23484
86. Zhou M, Li W, Wen Z, Sheng Y, Ren H, Dong H, et al. Macrophages enhance tumor-derived autophagosomes (DRibbles)-induced B cells activation by TLR4/MyD88 and CD40/CD40L. Exp Cell Res. 2015;331(2):320-30. https://doi.org/10.1016/j.yexcr.2014.10.015
87. Zavitsanou AM, Pillai R, Hao Y, Wu WL, Bartnicki E, Karakousi T, et al. KEAP1 mutation in lung adenocarcinoma promotes immune evasion and immunotherapy resistance. Cell Rep. 2023;42(11):113295. https://doi.org/10.1016/j.celrep.2023.113295
88. Luo M, Wang X, Wu S, Yang C, Su Q, Huang L, et al. A20 promotes colorectal cancer immune evasion by upregulating STC1 expression to block "eat-me" signal. Signal Transduct Target Ther. 2023;8(1):312. https://doi.org/10.1038/s41392-023-01545-x
89. Xiao D, Zeng T, Zhu W, Yu ZZ, Huang W, Yi H, et al. ANXA1 promotes tumor immune evasion by binding PARP1 and upregulating Stat3-induced expression of PD-L1 in multiple cancers. Cancer Immunol Res. 2023;11(10):1367-83. https://doi.org/10.1158/2326-6066.cir-22-0896
90. Wang J, Yang Y, Shao F, Meng Y, Guo D, He J, et al. Acetate reprogrammes tumour metabolism and promotes PD-L1 expression and immune evasion by upregulating c-Myc. Nat Metab. 2024;6(5):914-32. https://doi.org/10.1038/s42255-024-01037-4
91. Cheng M, Chen S, Li K, Wang G, Xiong G, Ling R, et al. CD276-dependent efferocytosis by tumor-associated macrophages promotes immune evasion in bladder cancer. Nat Commun. 2024;15(1):2818. https://doi.org/10.1038/s41467-024-46735-5
92. Zhang Y, Zeng L, Wang M, Yang Z, Zhang H, Gao L, et al. RIG-I promotes immune evasion of colon cancer by modulating PD-L1 ubiquitination. J Immunother Cancer. 2023;11(9):e007313. https://doi.org/10.1136/jitc-2023-007313
93. Liu Y, Peng Y, Du W, Yu C, Peng Z, Qin L, et al. PD-L1-mediated immune evasion in triple-negative breast cancer is linked to the loss of ZNF652. Cell Rep. 2023;42(11):113343. https://doi.org/10.1016/j.celrep.2023.113343
94. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581(7806):100-5. https://doi.org/10.1038/s41586-020-2229-5
95. Yamamoto K, Venida A, Perera RM, Kimmelman AC. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy. 2020;16(8):1524-5. https://doi.org/10.1080/15548627.2020.1769973
96. Zhan L, Zhang J, Wei B, Cao Y. Selective autophagy of NLRC5 promotes immune evasion of endometrial cancer. Autophagy. 2022;18(4):942-3. https://doi.org/10.1080/15548627.2022.2037119
97. Liu Y, Zhang H, Wang Z, Wu P, Gong W. 5-Hydroxytryptamine1a receptors on tumour cells induce immune evasion in lung adenocarcinoma patients with depression via autophagy/pSTAT3. Eur J Cancer. 2019;114:8-24. https://doi.org/10.1016/j.ejca.2019.03.017
98. Jin Y, Qiu J, Lu X, Li G. C-MYC inhibited ferroptosis and promoted immune evasion in ovarian cancer cells through NCOA4 mediated ferritin autophagy. Cells. 2022;11(24):4127. https://doi.org/10.3390/cells11244127
99. Fu Y, Mackowiak B, Feng D, Lu H, Guan Y, Lehner T, et al. MicroRNA-223 attenuates hepatocarcinogenesis by blocking hypoxia-driven angiogenesis and immunosuppression. Gut. 2023;72(10):1942-58. https://doi.org/10.1136/gutjnl-2022-327924
100. Wu L, Xue M, Lai S, Chen J, Lin Y, Ding N, et al. Hypoxia derived exosomes promote the proliferation and metastasis of colorectal cancer through the regulation of HIF-1α/miR-4299/ZBTB4. Life Sci. 2023;329:121872. https://doi.org/10.1016/j.lfs.2023.121872
101. Li M, Li L, Cheng X, Li L, Tu K. Hypoxia promotes the growth and metastasis of ovarian cancer cells by suppressing ferroptosis via upregulating SLC2A12. Exp Cell Res. 2023;433(2):113851. https://doi.org/10.1016/j.yexcr.2023.113851
102. Liu Y, Liu Y, He Y, Zhang N, Zhang S, Li Y, et al. Hypoxia-induced FUS-circTBC1D14 stress granules promote autophagy in TNBC. Adv Sci (Weinh). 2023;10(10):e2204988. https://doi.org/10.1002/advs.202204988
103. Feng X, Zhang H, Meng L, Song H, Zhou Q, Qu C, et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy. 2021;17(3):723-42. https://doi.org/10.1080/15548627.2020.1731266
104. Wang J, Dong Z, Sheng Z, Cai Y. Hypoxia-induced PVT1 promotes lung cancer chemoresistance to cisplatin by autophagy via PVT1/miR-140-3p/ATG5 axis. Cell Death Discov. 2022;8(1):104. https://doi.org/10.1038/s41420-022-00886-w
105. Li H, Zhang C, Zhang Q, Jia J, Wang X. BNIP3 enhances pancreatic cancer cell migration and proliferation via modulating autophagy under hypoxia. Heliyon. 2022;8(10):e11190. https://doi.org/10.1016/j.heliyon.2022.e11190
106. Kim TW, Lee HG. Apigenin induces autophagy and cell death by targeting EZH2 under hypoxia conditions in gastric cancer cells. Int J Mol Sci. 2021;22(24):13455. https://doi.org/10.3390/ijms222413455
107. Lai MC, Chang CM, Sun HS. Hypoxia induces autophagy through translational up-regulation of lysosomal proteins in human colon cancer cells. PLoS One. 2016;11(4):e0153627. https://doi.org/10.1371/journal.pone.0153627
108. Kim YS, Jeong YS, Bae GH, Kang JH, Lee M, Zabel BA, et al. CD200R high neutrophils with dysfunctional autophagy establish systemic immunosuppression by increasing regulatory T cells. Cell Mol Immunol. 2024;21(4):349-61. https://doi.org/10.1038/s41423-024-01136-y
109. Guo R, Zhao G, Bai G, Chen J, Han W, Cui N, et al. Depletion of mTOR ameliorates CD4+ T cell pyroptosis by promoting autophagy activity in septic mice. Int Immunopharmacol. 2023;124(Pt B):110964. https://doi.org/10.1016/j.intimp.2023.110964
110. Pang L, Guo S, Khan F, Dunterman M, Ali H, Liu Y, et al. Hypoxia-driven protease legumain promotes immunosuppression in glioblastoma. Cell Rep Med. 2023;4(11):101238. https://doi.org/10.1016/j.xcrm.2023.101238
111. Coleman MF, Kulkoyluoglu Cotul E, Pfeil AJ, Devericks EN, Safdar MH, Monteiro M, et al. Hypoxia-mediated repression of pyruvate carboxylase drives immunosuppression. Breast Cancer Res. 2024;26(1):96. https://doi.org/10.1186/s13058-024-01854-1
112. Liu Z, Sun L, Peng X, Liu S, Zhu Z, Huang C. An immunogenic cell death-related signature predicts prognosis and immunotherapy response in stomach adenocarcinoma. Apoptosis. 2023;28(11-12):1564-83. https://doi.org/10.1007/s10495-023-01879-5
113. Li Z, Lai X, Fu S, Ren L, Cai H, Zhang H, et al. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci (Weinh). 2022;9(22):e2201734. https://doi.org/10.1002/advs.202201734
114. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11(11):1013. https://doi.org/10.1038/s41419-020-03221-2
115. Wang J, Ma J, Tai Z, Li L, Zhang T, Cheng T, et al. Nanocarrier-mediated immunogenic cell death for melanoma treatment. Int J Nanomedicine. 2023;18:7149-72. https://doi.org/10.2147/ijn.s434582
116. Mondesir J, Ghisi M, Poillet L, Bossong RA, Kepp O, Kroemer G, et al. AMPK activation induces immunogenic cell death in AML. Blood Adv. 2023;7(24):7585-96. https://doi.org/10.1182/bloodadvances.2022009444
117. Ghosh S, Yang R, Duraki D, Zhu J, Kim JE, Jabeen M, et al. Plasma membrane channel TRPM4 mediates immunogenic therapy-induced necrosis. Cancer Res. 2023;83(18):3115-30. https://doi.org/10.1158/0008-5472.can-23-0157
118. Yu S, Xiao H, Ma L, Zhang J, Zhang J. Reinforcing the immunogenic cell death to enhance cancer immunotherapy efficacy. Biochim Biophys Acta Rev Cancer. 2023;1878(5):188946. https://doi.org/10.1016/j.bbcan.2023.188946
119. Pan H, Liu P, Zhao L, Pan Y, Mao M, Kroemer G, et al. Immunogenic cell stress and death in the treatment of cancer. Semin Cell Dev Biol. 2024;156:11-21. https://doi.org/10.1016/j.semcdb.2023.10.007
120. Meier P, Legrand AJ, Adam D, Silke J. Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity. Nat Rev Cancer. 2024;24(5):299-315. https://doi.org/10.1038/s41568-024-00674-x
121. Ishimwe N, Wei P, Wang M, Zhang H, Wang L, Jing M, et al. Autophagy impairment through lysosome dysfunction by brucine induces immunogenic cell death (ICD). Am J Chin Med. 2020;48(8):1915-40. https://doi.org/10.1142/s0192415x20500962
122. Xu L, Su B, Mo L, Zhao C, Zhao Z, Li H, et al. Norcantharidin induces immunogenic cell death of bladder cancer cells through promoting autophagy in acidic culture. Int J Mol Sci. 2022;23(7):3944. https://doi.org/10.3390/ijms23073944
123. Zhang S, Huang Y, Pi S, Chen H, Ye F, Wu C, et al. Autophagy-amplifying nanoparticles evoke immunogenic cell death combined with anti-PD-1/PD-L1 for residual tumors immunotherapy after RFA. J Nanobiotechnology. 2023;21(1):360. https://doi.org/10.1186/s12951-023-02067-y
124. Tran TH, Kao M, Liu HS, Hong YR, Su Y, Huang CYF. Repurposing thioridazine for inducing immunogenic cell death in colorectal cancer via eIF2α/ATF4/CHOP and secretory autophagy pathways. Cell Commun Signal. 2023;21(1):184. https://doi.org/10.1186/s12964-023-01190-5
125. Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, et al. ROS-induced autophagy in cancer cells assists in evasion from determinants of immunogenic cell death. Autophagy. 2013;9(9):1292-307. https://doi.org/10.4161/auto.25399
126. Ye T, Jiang K, Wei L, Barr MP, Xu Q, Zhang G, et al. Oncolytic Newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am J Cancer Res. 2018;8(8):1514-27. https://pubmed.ncbi.nlm.nih.gov/30210920
127. Lei Y, Zhang E, Bai L, Li Y. Autophagy in cancer immunotherapy. Cells. 2022;11(19):2996. https://doi.org/10.3390/cells11192996
128. Schauer IG, Zhang J, Xing Z, Guo X, Mercado-Uribe I, Sood AK, et al. Interleukin-1β promotes ovarian tumorigenesis through a p53/NF-κB-mediated inflammatory response in stromal fibroblasts. Neoplasia. 2013;15(4):409-20. https://doi.org/10.1593/neo.121228
129. Jiang S, Dupont N, Castillo EF, Deretic V. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun. 2013;5(5):471-9. https://doi.org/10.1159/000346707
130. Peral de Castro C, Jones SA, Ní Cheallaigh C, Hearnden CA, Williams L, Winter J, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol. 2012;189(8):4144-53. https://doi.org/10.4049/jimmunol.1201946
131. Liu E, Van Grol J, Subauste CS. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-γ production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect. 2015;17(4):275-84. https://doi.org/10.1016/j.micinf.2014.12.008
132. Sun K, Xu L, Jing Y, Han Z, Chen X, Cai C, et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017;388:198-207. https://doi.org/10.1016/j.canlet.2016.12.004
133. Kang R, Tang D, Lotze MT, Zeh HJ 3rd. Autophagy is required for IL-2-mediated fibroblast growth. Exp Cell Res. 2013;319(4):556-65. https://doi.org/10.1016/j.yexcr.2012.11.012
134. Qin B, Zhou Z, He J, Yan C, Ding S. IL-6 inhibits starvation-induced autophagy via the STAT3/Bcl-2 signaling pathway. Sci Rep. 2015;5:15701. https://doi.org/10.1038/srep15701
135. Linnemann AK, Blumer J, Marasco MR, Battiola TJ, Umhoefer HM, Han JY, et al. Interleukin 6 protects pancreatic β cells from apoptosis by stimulation of autophagy. FASEB J. 2017;31(9):4140-52. https://doi.org/10.1096/fj.201700061rr
136. Cho SH, Oh SY, Lane AP, Lee J, Oh MH, Lee S, et al. Regulation of nasal airway homeostasis and inflammation in mice by SHP-1 and Th2/Th1 signaling pathways. PLoS One. 2014;9(8):e103685. https://doi.org/10.1371/journal.pone.0103685
137. Schmeisser H, Bekisz J, Zoon KC. New function of type I IFN: induction of autophagy. J Interferon Cytokine Res. 2014;34(2):71-8. https://doi.org/10.1089/jir.2013.0128
138. Matsuzawa T, Kim BH, Shenoy AR, Kamitani S, Miyake M, Macmicking JD. IFN-γ elicits macrophage autophagy via the p38 MAPK signaling pathway. J Immunol. 2012;189(2):813-8. https://doi.org/10.4049/jimmunol.1102041
139. Ding Y, Kim SI, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol. 2014;25(12):2835-46. https://doi.org/10.1681/asn.2013101068
140. Buchser WJ, Laskow TC, Pavlik PJ, Lin HM, Lotze MT. Cell-mediated autophagy promotes cancer cell survival. Cancer Res. 2012;72(12):2970-9. https://doi.org/10.1158/0008-5472.can-11-3396
141. Khatib TO, Pedro BA, Bombin S, Matsuk VY, Robinson IE, Webster SF, et al. TGF-β1-mediated intercellular signaling fuels cooperative cellular invasion. Cell Rep. 2025;44(2):115315. https://doi.org/10.1016/j.celrep.2025.115315
142. Suzuki HI, Kiyono K, Miyazono K. Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy. 2010;6(5):645-7. https://doi.org/10.4161/auto.6.5.12046
143. Wang MX, Cheng XY, Jin M, Cao YL, Yang YP, Wang JD, et al. TNF compromises lysosome acidification and reduces α-synuclein degradation via autophagy in dopaminergic cells. Exp Neurol. 2015;271:112-21. https://doi.org/10.1016/j.expneurol.2015.05.008
144. Ullio C, Brunk UT, Urani C, Melchioretto P, Bonelli G, Baccino FM, et al. Autophagy of metallothioneins prevents TNF-induced oxidative stress and toxicity in hepatoma cells. Autophagy. 2015;11(12):2184-98. https://doi.org/10.1080/15548627.2015.1106662
145. Qin R, Ren W, Ya G, Wang B, He J, Ren S, et al. Role of chemokines in the crosstalk between tumor and tumor-associated macrophages. Clin Exp Med. 2023;23(5):1359-73. https://doi.org/10.1007/s10238-022-00888-z
146. Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44(3):439-49. https://doi.org/10.1016/j.immuni.2016.02.024
147. Aehnlich P, Powell RM, Peeters MJW, Rahbech A, Straten PT. TAM receptor inhibition-implications for cancer and the immune system. Cancers (Basel). 2021;13(6):1195. https://doi.org/10.3390/cancers13061195
148. Zhang L, Jiang Y, Zhang G, Wei S. The diversity and dynamics of tumor-associated macrophages in recurrent glioblastoma. Front Immunol. 2023;14:1238233. https://doi.org/10.3389/fimmu.2023.1238233
149. Li Y, Wang R, Gao Q. The roles and targeting of tumor-associated macrophages. Front Biosci (Landmark Ed). 2023;28(9):207. https://doi.org/10.31083/j.fbl2809207
150. Wang YN, Wang YY, Wang J, Bai WJ, Miao NJ, Wang J. Vinblastine resets tumor-associated macrophages toward M1 phenotype and promotes antitumor immune response. J Immunother Cancer. 2023;11(8):e007253. https://doi.org/10.1136/jitc-2023-007253
151. Zhang G, Gao Z, Guo X, Ma R, Wang X, Zhou P, et al. CAP2 promotes gastric cancer metastasis by mediating the interaction between tumor cells and tumor-associated macrophages. J Clin Invest. 2023;133(21):e166224. https://doi.org/10.1172/jci166224
152. Do MH, Shi W, Ji L, Ladewig E, Zhang X, Srivastava RM, et al. Reprogramming tumor-associated macrophages to outcompete endovascular endothelial progenitor cells and suppress tumor neoangiogenesis. Immunity. 2023;56(11):2555-2569.e5. https://doi.org/10.1016/j.immuni.2023.10.010
153. Rohila D, Park IH, Pham TV, Weitz J, Hurtado de Mendoza T, Madheswaran S, et al. Syk inhibition reprograms tumor-associated macrophages and overcomes gemcitabine-induced immunosuppression in pancreatic ductal adenocarcinoma. Cancer Res. 2023;83(16):2675-89. https://doi.org/10.1158/0008-5472.can-22-3645
154. Wang S, Yan W, Kong L, Zuo S, Wu J, Zhu C, et al. Oncolytic viruses engineered to enforce cholesterol efflux restore tumor-associated macrophage phagocytosis and anti-tumor immunity in glioblastoma. Nat Commun. 2023;14(1):4367. https://doi.org/10.1038/s41467-023-39683-z
155. Yuan D, Hu J, Ju X, Putz EM, Zheng S, Koda S, et al. NMDAR antagonists suppress tumor progression by regulating tumor-associated macrophages. Proc Natl Acad Sci U S A. 2023;120(47):e2302126120. https://doi.org/10.1073/pnas.2302126120
156. Huang J, Pan H, Sun J, Wu J, Xuan Q, Wang J, et al. TMEM147 aggravates the progression of HCC by modulating cholesterol homeostasis, suppressing ferroptosis, and promoting the M2 polarization of tumor-associated macrophages. J Exp Clin Cancer Res. 2023;42(1):286. https://doi.org/10.1186/s13046-023-02865-0
157. Zhang S, Peng W, Wang H, Xiang X, Ye L, Wei X, et al. C1q+ tumor-associated macrophages contribute to immunosuppression through fatty acid metabolic reprogramming in malignant pleural effusion. J Immunother Cancer. 2023;11(8):e007441. https://doi.org/10.1136/jitc-2023-007441
158. Wang L, Guo W, Guo Z, Yu J, Tan J, Simons DL, et al. PD-L1-expressing tumor-associated macrophages are immunostimulatory and associate with good clinical outcome in human breast cancer. Cell Rep Med. 2024;5(2):101420. https://doi.org/10.1016/j.xcrm.2024.101420
159. Cai Z, Li W, Hager S, Wilson JL, Afjehi-Sadat L, Heiss EH, et al. Targeting PHGDH reverses the immunosuppressive phenotype of tumor-associated macrophages through α-ketoglutarate and mTORC1 signaling. Cell Mol Immunol. 2024;21(5):448-65. https://doi.org/10.1038/s41423-024-01134-0
160. Yang SL, Tan HX, Niu TT, Liu YK, Gu CJ, Li DJ, et al. The IFN-γ–IDO1–kynurenine pathway-induced autophagy in cervical cancer cells promotes phagocytosis of macrophages. Int J Biol Sci. 2021;17(1):339-52. https://doi.org/10.7150/ijbs.51241
161. Li Z, Fu WJ, Chen XQ, Wang S, Deng RS, Tang XP, et al. Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J Exp Clin Cancer Res. 2022;41(1):74. https://doi.org/10.1186/s13046-022-02291-8
162. Sanjurjo L, Aran G, Téllez É, Amézaga N, Armengol C, López D, et al. CD5L promotes M2 macrophage polarization through autophagy-mediated upregulation of ID3. Front Immunol. 2018;9:480. https://doi.org/10.3389/fimmu.2018.00480
163. Cotzomi-Ortega I, Nieto-Yañez O, Juárez-Avelar I, Rojas-Sanchez G, Montes-Alvarado JB, Reyes-Leyva J. Autophagy inhibition in breast cancer cells induces ROS-mediated MIF expression and M1 macrophage polarization. Cell Signal. 2021;86:110075. https://doi.org/10.1016/j.cellsig.2021.110075
164. Shan M, Qin J, Jin F, Han X, Guan H, Li X, et al. Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mTOR signaling pathway. Free Radic Biol Med. 2017;110:432-43. https://doi.org/10.1016/j.freeradbiomed.2017.05.021
165. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582-98. https://doi.org/10.1038/nrc.2016.73
166. Erez N, Truitt M, Olson P, Tuttleton Arron S, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell. 2010;17(2):135-47. https://doi.org/10.1016/j.ccr.2009.12.041
167. Bai J, Liu T, Tu B, Yuan M, Shu Z, Fan M, et al. Autophagy loss impedes cancer-associated fibroblast activation via downregulating proline biosynthesis. Autophagy. 2023;19(2):632-43. https://doi.org/10.1080/15548627.2022.2093026
168. Yuan M, Tu B, Li H, Pang H, Zhang N, Fan M, et al. Cancer-associated fibroblasts employ NUFIP1-dependent autophagy to secrete nucleosides and support pancreatic tumor growth. Nat Cancer. 2022;3(8):945-60. https://doi.org/10.1038/s43018-022-00426-6
169. Xi L, Peng M, Liu S, Liu Y, Wan X, Hou Y, et al. Hypoxia-stimulated ATM activation regulates autophagy-associated exosome release from cancer-associated fibroblasts to promote cancer cell invasion. J Extracell Vesicles. 2021;10(11):e12146. https://doi.org/10.1002/jev2.12146
170. Dong D, Yao Y, Song J, Sun L, Zhang G. Cancer-associated fibroblasts regulate bladder cancer invasion and metabolic phenotypes through autophagy. Dis Markers. 2021;2021:6645220. https://doi.org/10.1155/2021/6645220
171. Wang Y, Gan G, Wang B, Wu J, Cao Y, Zhu D, et al. Cancer-associated fibroblasts promote irradiated cancer cell recovery through autophagy. EBioMedicine. 2017;17:45-56. https://doi.org/10.1016/j.ebiom.2017.02.019
172. Wang M, Zhang J, Huang Y, Ji S, Shao G, Feng S, et al. Cancer-associated fibroblasts autophagy enhances progression of triple-negative breast cancer cells. Med Sci Monit. 2017;23:3904-12. https://doi.org/10.12659/msm.902870
173. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480(7378):480-9. https://doi.org/10.1038/nature10673
174. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265-77. https://doi.org/10.1038/nrc3258
175. Xiong H, Chen Z, Lin B, Xie B, Liu X, Chen C, et al. Naringenin regulates FKBP4/NR3C1/NRF2 axis in autophagy and proliferation of breast cancer and differentiation and maturation of dendritic cell. Front Immunol. 2022;12:745111. https://doi.org/10.3389/fimmu.2021.745111
176. Liu Y, Yin Z, Lu P, Ma Y, Luo B, Xiang L, et al. Lung carcinoma cells secrete exosomal MALAT1 to inhibit dendritic cell phagocytosis, inflammatory response, costimulatory molecule expression and promote dendritic cell autophagy via AKT/mTOR pathway. Onco Targets Ther. 2020;13:10693-705. https://doi.org/10.2147/ott.s256669
177. Zamame Ramirez JA, Romagnoli GG, Falasco BF, Gorgulho CM, Fogolin CS, Dos Santos DC. Blocking drug-induced autophagy with chloroquine in HCT-116 colon cancer cells enhances DC maturation and T cell responses induced by tumor cell lysate. Int Immunopharmacol. 2020;84:106495. https://doi.org/10.1016/j.intimp.2020.106495
178. Wang Y, Lin YX, Wang J, Qiao SL, Liu YY, Dong WQ, et al. In situ manipulation of dendritic cells by an autophagy-regulative nanoactivator enables effective cancer immunotherapy. ACS Nano. 2019;13(7):7568-77. https://doi.org/10.1021/acsnano.9b00143
179. Shen T, Zhu W, Yang L, Liu L, Jin R, Duan J, et al. Lactosylated N-alkyl polyethylenimine coated iron oxide nanoparticles induced autophagy in mouse dendritic cells. Regen Biomater. 2018;5(3):141-9. https://doi.org/10.1093/rb/rbx032
180. Mace EM. Human natural killer cells: form, function, and development. J Allergy Clin Immunol. 2023;151(2):371-85. https://doi.org/10.1016/j.jaci.2022.09.022
181. Raulet DH, Guerra N. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol. 2009;9(8):568-80. https://doi.org/10.1038/nri2604
182. Zhang J, Zhou L, Xiang JD, Jin CS, Li MQ, He YY. Artesunate-induced ATG5-related autophagy enhances the cytotoxicity of NK92 cells on endometrial cancer cells via interactions between CD155 and CD226/TIGIT. Int Immunopharmacol. 2021;97:107705. https://doi.org/10.1016/j.intimp.2021.107705
183. Li ZL, Zhang HL, Huang Y, Huang JH, Sun P, Zhou NN, et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat Commun. 2020;11(1):3806. https://doi.org/10.1038/s41467-020-17395-y
184. Tang L, Zhang H, Zhou F, Wei Q, Du M, Wu J, et al. Targeting autophagy overcomes cancer-intrinsic resistance to CAR-T immunotherapy in B-cell malignancies. Cancer Commun (Lond). 2024;44(3):408-32. https://doi.org/10.1002/cac2.12525
185. Buono R, Tucci J, Cutri R, Guidi N, Mangul S, Raucci F, et al. Fasting-mimicking diet inhibits autophagy and synergizes with chemotherapy to promote T-cell-dependent leukemia-free survival. Cancers (Basel). 2023;15(24):5870. https://doi.org/10.3390/cancers15245870
186. Young TM, Reyes C, Pasnikowski E, Castanaro C, Wong C, Decker CE, et al. Autophagy protects tumors from T cell-mediated cytotoxicity via inhibition of TNFα-induced apoptosis. Sci Immunol. 2020;5(54):eabb9561. https://doi.org/10.1126/sciimmunol.abb9561
187. Nunn K, Guo JY. Transient systemic autophagy ablation irreversibly inhibits lung tumor cell metabolism and promotes T-cell mediated tumor killing. Autophagy. 2023;19(6):1879-81. https://doi.org/10.1080/15548627.2022.2141534
188. Xia H, Wang W, Crespo J, Kryczek I, Li W, Wei S, et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naïve T cell apoptosis and affects tumor immunity. Sci Immunol. 2017;2(17):eaan4631. https://doi.org/10.1126/sciimmunol.aan4631
189. Dikiy S, Rudensky AY. Principles of regulatory T cell function. Immunity. 2023;56(2):240-55. https://doi.org/10.1016/j.immuni.2023.01.004
190. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109-18. https://doi.org/10.1038/cr.2016.151
191. Yamazaki S, Bonito AJ, Spisek R, Dhodapkar M, Inaba K, Steinman RM. Dendritic cells are specialized accessory cells along with TGF-β for the differentiation of Foxp3+ CD4+ regulatory T cells from peripheral Foxp3 precursors. Blood. 2007;110(13):4293-302. https://doi.org/10.1182/blood-2007-05-088831
192. Swatler J, Ju YJ, Anderson AC, Lugli E. Tumors recycle glucocorticoids to drive Treg-mediated immunosuppression. J Clin Invest. 2023;133(18):e173141. https://doi.org/10.1172/jci173141
193. Wang Q, Guo W, Niu L, Zhou Y, Wang Z, Chen J, et al. 3D-hUMSCs exosomes ameliorate vitiligo by simultaneously potentiating Treg cells-mediated immunosuppression and suppressing oxidative stress-induced melanocyte damage. Adv Sci (Weinh). 2024;11(31):e2404064. https://doi.org/10.1002/advs.202404064
194. Liu ZY, Lin XH, Guo HY, Shi X, Zhang DY, Sun JL, et al. Multi-omics profiling identifies aldehyde dehydrogenase 2 as a critical mediator in the crosstalk between Treg-mediated immunosuppression microenvironment and hepatocellular carcinoma. Int J Biol Sci. 2024;20(7):2763-78. https://doi.org/10.7150/ijbs.93075
195. Gerner MC, Ziegler LS, Schmidt RLJ, Krenn M, Zimprich F, Uyanik-Unal K, et al. The TGF-β/SOX4 axis and ROS-driven autophagy co-mediate CD39 expression in regulatory T-cells. FASEB J. 2020;34(6):8367-84. https://doi.org/10.1096/fj.201902664
196. Lasser SA, Ozbay Kurt FG, Arkhypov I, Utikal J, Umansky V. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat Rev Clin Oncol. 2024;21(2):147-64. https://doi.org/10.1038/s41571-023-00846-y
197. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3-8. https://doi.org/10.1158/2326-6066.cir-16-0297
198. Tian X, Wang T, Shen H, Wang S. Tumor microenvironment, histone modifications, and myeloid-derived suppressor cells. Cytokine Growth Factor Rev. 2023;74:108-21. https://doi.org/10.1016/j.cytogfr.2023.08.002
199. Okła K. Myeloid-derived suppressor cells (MDSCs) in ovarian cancer-looking back and forward. Cells. 2023;12(14):1912. https://doi.org/10.3390/cells12141912
200. Nguyen KA, DePledge LN, Bian L, Ke Y, Samedi V, Berning AA, et al. Polymorphonuclear myeloid-derived suppressor cells and phosphatidylinositol-3 kinase gamma are critical to tobacco-mimicking oral carcinogenesis in mice. J Immunother Cancer. 2023;11(9):e007110. https://doi.org/10.1136/jitc-2023-007110
201. Calderon JJ, Prieto K, Lasso P, Fiorentino S, Barreto A. Modulation of myeloid-derived suppressor cells in the tumor microenvironment by natural products. Arch Immunol Ther Exp (Warsz). 2023;71(1):17. https://doi.org/10.1007/s00005-023-00681-0
202. van Wigcheren GF, Cuenca-Escalona J, Stelloo S, Brake J, Peeters E, Horrevorts SK, Frölich S, et al. Myeloid-derived suppressor cells and tolerogenic dendritic cells are distinctively induced by PI3K and Wnt signaling pathways. J Biol Chem. 2023;299(11):105276. https://doi.org/10.1016/j.jbc.2023.105276
203. Dong G, Si C, Zhang Q, Yan F, Li C, Zhang H, et al. Autophagy regulates accumulation and functional activity of granulocytic myeloid-derived suppressor cells via STAT3 signaling in endotoxin shock. Biochim Biophys Acta Mol Basis Dis. 2017;1863(11):2796-807. https://doi.org/10.1016/j.bbadis.2017.08.005
204. Xia C, Li M, Ran G, Wang X, Lu Z, Li T, et al. Redox-responsive nanoassembly restrained myeloid-derived suppressor cells recruitment through autophagy-involved lactate dehydrogenase A silencing for enhanced cancer immunochemotherapy. J Control Release. 2021;335:557-74. https://doi.org/10.1016/j.jconrel.2021.05.034
205. Wu JS, Li L, Wang SS, Pang X, Wu JB, Sheng SR, et al. Autophagy is positively associated with the accumulation of myeloid-derived suppressor cells in 4-nitroquinoline-1-oxide-induced oral cancer. Oncol Rep. 2018;40(6):3381-91. https://doi.org/10.3892/or.2018.6747
206. Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest. 2018;128(9):3840-52. https://doi.org/10.1172/jci120888
207. Zhang W, Li X, Jiang M, Ji C, Chen G, Zhang Q, et al. SOCS3 deficiency-dependent autophagy repression promotes the survival of early-stage myeloid-derived suppressor cells in breast cancer by activating the Wnt/mTOR pathway. J Leukoc Biol. 2023;113(5):445-60. https://doi.org/10.1093/jleuko/qiad020
208. Parker KH, Horn LA, Ostrand-Rosenberg S. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J Leukoc Biol. 2016;100(3):463-70. https://doi.org/10.1189/jlb.3hi0715-305r
209. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16(7):431-46. https://doi.org/10.1038/nrc.2016.52
210. Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2007;104(51):20262-7. https://doi.org/10.1073/pnas.0706438104
211. Wu Y, Liu Q, Xie Y, Zhu J, Zhang S, Ge Y, et al. MUC16 stimulates neutrophils to an inflammatory and immunosuppressive phenotype in ovarian cancer. J Ovarian Res. 2023;16(1):181. https://doi.org/10.1186/s13048-023-01207-0
212. Lu Z, Wang X, Feng J, Chai W, Wang W, Wang Q, et al. Intratumoral CXCR4^hi neutrophils display ferroptotic and immunosuppressive signatures in hepatoblastoma. Front Immunol. 2024;15:1363454. https://doi.org/10.3389/fimmu.2024.1363454
213. Kwantwi LB. Interplay between tumor-derived factors and tumor-associated neutrophils: opportunities for therapeutic interventions in cancer. Clin Transl Oncol. 2023;25(7):1963-76. https://doi.org/10.1007/s12094-023-03100-0
214. Liao C, Luo S, Liu X, Zhang L, Xie P, Zhou W, et al. Siglec-F+ neutrophils in the spleen induce immunosuppression following acute infection. Theranostics. 2024;14(6):2589-604. https://doi.org/10.7150/thno.93812
215. Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, et al. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol. 2015;62(1):131-9. https://doi.org/10.1016/j.jhep.2014.08.023
Declarations
Funding Statement: This work was supported by the National Natural Science Foundation of China (NSFC Grant No. 82472182).
Declaration of Competing Interest: The authors declare no financial or personal relationships with other individuals or organizations that could inappropriately influence or bias the content of this work. All authors have read the final version of the manuscript and confirm that there are no competing interests.
Consent for publication: All authors have approved the final version of the manuscript.
Use of Artificial Intelligence (AI) Disclosure: This article was written by human contributors. Artificial intelligence-based tools were used to improve grammar, language, and readability without affecting the article's scientific content, data interpretation, or conclusions. The authors reviewed and verified all content to ensure its accuracy and integrity.
Dual Use Research of Concern (DURC): “The authors confirm that this research does not constitute DURC as defined by the U.S. Government Policy for Oversight of Life Sciences DURC.”
Data Availability Statements (DAS): “No datasets were generated or analyzed in the current study.”
Ethics approval and consent to participate: Not applicable, as this study did not involve the conduct of research.
Authors’ affiliations
1 School of Pharmacy and Bioengineering, Chongqing University of Technology, Chongqing 400054, PR China
2 Department of Anesthesiology, The Second Affiliated Hospital of Chongqing Medical University, Chongqing 400010, P.R. China
3 Department of Neurology, Southwest Hospital, Army Medical University, Chongqing 400038, P.R. China
*These authors contribute equally to this article.
#Correspondance: Jun Hu, Email: hujun@tmmu.edu.cn
Chenyang Duan, Email: duanchenyang1991@cqmu.edu.cn
CRediT authorship contribution statement
LYC and TXX were involved in study conceptualization, literature review, and manuscript drafting. RS contributed to the literature collection and manuscript revision. DCY and HJ contributed to conceptualization, supervision, and critical revision of the manuscript. The authors read and approved the final manuscript.
ORCID ID
Jun Hu: https://orcid.org/0000-0001-9355-8318
How to cite
Liao Y, Tan X, Ren S, Duan C, Hu J. Autophagy-Driven Cancer Immunotherapy via Tumor Immune Ecosystem Remodeling. Cancer Biome and Targeted Therapy. Published online 2025 Dec.